---
title: "Clasificadores Supervisados: Experimento Práctico"
author: "Diego Villalba"
date: today
lang: es
format:
html:
toc: true
toc-depth: 3
toc-title: "Contenido"
number-sections: true
code-fold: false
code-tools: true
code-summary: "Mostrar código"
fig-align: center
theme: cosmo
highlight-style: github
smooth-scroll: true
pdf:
toc: true
toc-depth: 3
number-sections: true
documentclass: scrbook
papersize: letter
fontsize: 11pt
geometry: margin=2.5cm
keep-tex: false
execute:
echo: true
warning: false
message: false
cache: false
bibliography: references_example.bib
crossref:
fig-title: "Figura"
tbl-title: "Tabla"
eq-prefix: "Ec."
---
```{python}
#| echo: false
#| label: setup-capitulo
import numpy as np
import pandas as pd
import matplotlib.pyplot as plt
import warnings
warnings.filterwarnings("ignore")
import matplotlib
matplotlib.rcParams.update({
"figure.dpi": 100,
"axes.spines.top": False,
"axes.spines.right": False,
"font.size": 11,
})
SEED = 42
np.random.seed(SEED)
```
# Introducción
La clasificación supervisada es una de las tareas centrales del aprendizaje automático. Dado un conjunto de observaciones etiquetadas $\{(\mathbf{x}_i, y_i)\}_{i=1}^{n}$, donde $\mathbf{x}_i \in \mathbb{R}^d$ es el vector de características y $y_i \in \{0, 1, \ldots, K-1\}$ es la clase objetivo, el problema de clasificación consiste en aprender una función $f: \mathbb{R}^d \to \mathcal{Y}$ que generalice correctamente a nuevas observaciones no vistas durante el entrenamiento.
Este capítulo no busca derivar los fundamentos teóricos de cada algoritmo desde cero, sino mostrar cómo se diseña, ejecuta y evalúa un experimento de clasificación real. El énfasis está en los retos prácticos: cómo preparar los datos, cómo evitar errores comunes, cómo comparar modelos de forma justa y cómo interpretar los resultados en función del contexto del problema.
---
# Dataset: Breast Cancer Wisconsin
El dataset Breast Cancer Wisconsin [@wolberg1995breast] contiene 569 muestras de biopsias de tejido mamario, cada una descrita por 30 variables numéricas derivadas de imágenes de células. La variable objetivo indica si el tumor es maligno (0) o benigno (1). Este dataset tiene varias propiedades didácticas deseables:
- Está disponible directamente en scikit-learn, sin necesidad de descarga externa
- Todas las variables son numéricas, lo que simplifica el preprocesamiento
- El contexto médico es motivador: los errores tienen consecuencias asimétricas
- El desbalance moderado (37% maligno, 63% benigno) permite discutir la elección de métricas
```{python}
#| label: carga-capitulo
#| code-fold: false
from sklearn.datasets import load_breast_cancer
data = load_breast_cancer()
X = pd.DataFrame(data.data, columns=data.feature_names)
y = pd.Series(data.target, name="target")
print(f"Forma del dataset: {X.shape}")
print(f"Distribución de clases:")
print(y.value_counts().rename({0: "Maligno (0)", 1: "Benigno (1)"}))
```
Un aspecto importante es identificar correctamente la clase positiva. Por convención en scikit-learn, `target=1` corresponde a benigno y `target=0` a maligno. Cuando se reportan métricas como precision y recall, es necesario especificar con respecto a qué clase se calculan, ya que los valores pueden cambiar sustancialmente dependiendo de esta elección.
---
# Flujo general de un experimento de clasificación
Un experimento bien diseñado sigue estos pasos:
1. **Entender el problema** y la variable objetivo
2. **Explorar y preparar los datos**
3. **Dividir correctamente** en entrenamiento y prueba
4. **Elegir modelos** y configurar hiperparámetros iniciales
5. **Entrenar** usando únicamente los datos de entrenamiento
6. **Evaluar** con métricas coherentes con el objetivo del problema
7. **Comparar modelos** con la misma partición de datos
8. **Ajustar thresholds** si el contexto lo requiere
9. **Interpretar resultados** y reconocer limitaciones
10. **Documentar** el flujo para que sea reproducible
Cada uno de estos pasos involucra decisiones que afectan la validez del experimento. En las secciones siguientes se desarrolla cada uno con detalle.
---
# Preparación de datos
## Separación entrenamiento/prueba
La regla fundamental es que el conjunto de prueba no debe usarse de ninguna manera durante el diseño, el entrenamiento o el ajuste de los modelos. Usar información del test set para tomar decisiones de diseño introduce **data leakage**, que produce una evaluación optimista que no se replicará en datos reales.
```{python}
#| label: split-capitulo
#| code-fold: false
from sklearn.model_selection import train_test_split
X_train, X_test, y_train, y_test = train_test_split(
X, y,
test_size=0.2,
random_state=SEED,
stratify=y
)
print(f"Entrenamiento: {X_train.shape[0]} muestras")
print(f"Prueba: {X_test.shape[0]} muestras")
```
El parámetro `stratify=y` garantiza que la proporción de clases se mantiene tanto en train como en test, lo que es especialmente importante con datasets desbalanceados.
## Pipelines para preprocesamiento sin leakage
La forma correcta de aplicar transformaciones como el escalamiento es dentro de un `Pipeline` de scikit-learn [@pedregosa2011sklearn]. Un pipeline asegura que el ajuste de cualquier transformación (por ejemplo, `StandardScaler`) se realiza únicamente sobre los datos de entrenamiento, y la misma transformación ya ajustada se aplica automáticamente a los datos de prueba.
```{python}
#| label: pipeline-capitulo
from sklearn.pipeline import Pipeline
from sklearn.preprocessing import StandardScaler
from sklearn.linear_model import LogisticRegression
pipe = Pipeline([
("scaler", StandardScaler()),
("clf", LogisticRegression(max_iter=1000, random_state=SEED))
])
pipe.fit(X_train, y_train)
print(f"Accuracy en test: {pipe.score(X_test, y_test):.4f}")
```
El error clásico de data leakage por escalamiento ocurre cuando se transforma todo el dataset antes del split:
```python
# ✗ MAL: StandardScaler ajustado sobre X completo (incluye test)
scaler = StandardScaler()
X_scaled = scaler.fit_transform(X)
X_train_s, X_test_s = train_test_split(X_scaled, ...)
```
## Validación cruzada
Cuando el tamaño del dataset es limitado, la evaluación con una sola partición train/test puede ser inestable. La validación cruzada $k$-fold [@kohavi1995cross] divide el conjunto de entrenamiento en $k$ partes (*folds*), entrena $k$ veces usando $k-1$ folds y evalúa en el fold restante, promediando los resultados.
```{python}
#| label: cv-capitulo
from sklearn.model_selection import cross_val_score
from sklearn.ensemble import RandomForestClassifier
pipe_rf = Pipeline([
("clf", RandomForestClassifier(n_estimators=100, random_state=SEED))
])
scores = cross_val_score(pipe_rf, X_train, y_train, cv=5, scoring="f1")
print(f"F1 por fold: {np.round(scores, 4)}")
print(f"Media: {scores.mean():.4f} ± {scores.std():.4f}")
```
La validación cruzada se usa para seleccionar hiperparámetros, no para evaluar el rendimiento final: el test set sigue siendo el árbitro final del rendimiento.
---
# Clasificadores: intuición, sesgos y hiperparámetros
## Regresión Logística (baseline)
La regresión logística modela la probabilidad de pertenencia a la clase positiva mediante una función sigmoide aplicada a una combinación lineal de las variables:
$$P(y=1 \mid \mathbf{x}) = \sigma(\mathbf{w}^\top \mathbf{x} + b) = \frac{1}{1 + e^{-(\mathbf{w}^\top \mathbf{x} + b)}}$$
Es el baseline lineal interpretable por excelencia. Sus coeficientes se pueden inspeccionar directamente para entender la contribución de cada variable. **Requiere escalamiento** para que el optimizador converja adecuadamente y para que los coeficientes sean comparables.
Hiperparámetros principales:
- `C`: inverso de la regularización (mayor C → menos regularización)
- `penalty`: tipo de regularización (`l1`, `l2`, `elasticnet`)
- `max_iter`: número máximo de iteraciones del optimizador
## Árboles de decisión: CART e ID3
### CART [@breiman1984cart]
CART (*Classification and Regression Trees*) construye árboles binarios mediante particiones recursivas del espacio de características. En cada nodo, selecciona el atributo y el umbral de corte que minimizan la impureza del nodo resultante. La impureza Gini se define como:
$$G = 1 - \sum_{k=0}^{K-1} p_k^2$$
donde $p_k$ es la proporción de muestras de la clase $k$ en el nodo.
### ID3 [@quinlan1986id3]
ID3 (*Iterative Dichotomiser 3*) utiliza la ganancia de información para seleccionar el atributo de partición:
$$\text{Ganancia}(S, A) = H(S) - \sum_{v \in \text{Valores}(A)} \frac{|S_v|}{|S|} H(S_v)$$
donde $H(S) = -\sum_k p_k \log_2 p_k$ es la entropía del conjunto $S$. En scikit-learn, ID3 se aproxima con `criterion="entropy"` en `DecisionTreeClassifier`.
En la práctica, la diferencia entre Gini y entropía es pequeña en la mayoría de los datasets. La entropía tiende a producir árboles ligeramente más profundos en datasets con muchas categorías nominales.
**Hiperparámetros principales de los árboles:**
- `max_depth`: profundidad máxima (controla sobreajuste)
- `min_samples_split`: mínimo de muestras para dividir un nodo
- `min_samples_leaf`: mínimo de muestras en una hoja
```{python}
#| label: cart-hiperparametros
from sklearn.tree import DecisionTreeClassifier
from sklearn.metrics import f1_score
for criterion in ["gini", "entropy"]:
for depth in [3, 5, None]:
pipe = Pipeline([
("clf", DecisionTreeClassifier(
criterion=criterion, max_depth=depth, random_state=SEED
))
])
pipe.fit(X_train, y_train)
f1 = f1_score(y_test, pipe.predict(X_test))
print(f"criterion={criterion:7s} | max_depth={str(depth):5s} | F1={f1:.4f}")
```
## Random Forest [@breiman2001random]
Random Forest es un ensamble de árboles de decisión construidos en paralelo mediante *bootstrap aggregating* (bagging). Cada árbol se entrena sobre una muestra con reemplazo del conjunto de entrenamiento, y en cada nodo solo se considera un subconjunto aleatorio de variables (`max_features`). El resultado final es la moda o el promedio de las predicciones de todos los árboles.
La aleatorización reduce la varianza del modelo compuesto, ya que los árboles individuales son decorrelacionados entre sí. Random Forest no requiere escalamiento y es relativamente robusto a hiperparámetros.
**Hiperparámetros principales:**
- `n_estimators`: número de árboles (más árboles → menor varianza, pero mayor costo)
- `max_features`: fracción de variables consideradas por árbol (`sqrt`, `log2`, fracción)
- `max_depth`: profundidad máxima de cada árbol
- `min_samples_leaf`: mínimo de muestras en hojas
```{python}
#| label: rf-capitulo
from sklearn.ensemble import RandomForestClassifier
from sklearn.metrics import roc_auc_score
pipe_rf = Pipeline([
("clf", RandomForestClassifier(
n_estimators=100, max_depth=None, random_state=SEED
))
])
pipe_rf.fit(X_train, y_train)
f1 = f1_score(y_test, pipe_rf.predict(X_test))
auc = roc_auc_score(y_test, pipe_rf.predict_proba(X_test)[:,1])
print(f"Random Forest — F1={f1:.4f} AUC={auc:.4f}")
```
## Gradient Boosting [@friedman2001gradient]
Gradient Boosting construye un ensamble de modelos de forma **secuencial**. Cada modelo nuevo se entrena para corregir los errores (residuales) del ensamble acumulado hasta ese momento. El algoritmo puede verse como un descenso por gradiente en el espacio de funciones, donde la función de pérdida (por ejemplo, log-loss para clasificación) guía la dirección de corrección.
A diferencia de Random Forest, Gradient Boosting es sensible a la tasa de aprendizaje (`learning_rate`) y a la profundidad de los árboles individuales. Una tasa de aprendizaje baja requiere más estimadores para alcanzar la misma capacidad, pero suele generalizar mejor.
XGBoost [@chen2016xgboost] es una implementación optimizada de Gradient Boosting que añade regularización explícita, manejo eficiente de valores faltantes y aceleración mediante paralelización de la construcción de árboles.
**Hiperparámetros principales:**
- `n_estimators`: número de árboles secuenciales
- `learning_rate`: paso del descenso por gradiente (típicamente 0.01–0.3)
- `max_depth`: profundidad de cada árbol (típicamente 3–6)
- `subsample`: fracción del dataset usada por árbol
```{python}
#| label: gb-capitulo
from sklearn.ensemble import GradientBoostingClassifier
pipe_gb = Pipeline([
("clf", GradientBoostingClassifier(
n_estimators=100, learning_rate=0.1,
max_depth=3, random_state=SEED
))
])
pipe_gb.fit(X_train, y_train)
f1 = f1_score(y_test, pipe_gb.predict(X_test))
auc = roc_auc_score(y_test, pipe_gb.predict_proba(X_test)[:,1])
print(f"Gradient Boosting — F1={f1:.4f} AUC={auc:.4f}")
```
## Red neuronal multicapa (MLP)
Una red neuronal multicapa (MLP, *Multilayer Perceptron*) aproxima funciones mediante composición de transformaciones afines y no lineales [@bishop2006prml; @goodfellow2016deep]:
$$\mathbf{h}^{(l)} = \sigma\!\left(\mathbf{W}^{(l)} \mathbf{h}^{(l-1)} + \mathbf{b}^{(l)}\right)$$
donde $\sigma$ es la función de activación (ReLU, tanh, sigmoide), $\mathbf{W}^{(l)}$ son los pesos de la capa $l$ y $\mathbf{h}^{(0)} = \mathbf{x}$ es la entrada. Los pesos se optimizan mediante retropropagación del gradiente.
Las redes neuronales son modelos muy flexibles, pero son más sensibles que los árboles a los hiperparámetros y al escalamiento de las variables de entrada. Si las entradas tienen escalas muy distintas, los gradientes pueden ser inestables y el optimizador converge lentamente o a mínimos deficientes.
**Hiperparámetros principales en MLPClassifier:**
- `hidden_layer_sizes`: arquitectura de capas ocultas (p.ej., `(64, 32)`)
- `activation`: función de activación (`relu`, `tanh`)
- `alpha`: regularización L2
- `max_iter`: épocas máximas de entrenamiento
```{python}
#| label: mlp-capitulo
from sklearn.neural_network import MLPClassifier
# Con escalamiento (correcto)
pipe_mlp = Pipeline([
("scaler", StandardScaler()),
("clf", MLPClassifier(
hidden_layer_sizes=(64, 32),
max_iter=300, random_state=SEED
))
])
pipe_mlp.fit(X_train, y_train)
f1 = f1_score(y_test, pipe_mlp.predict(X_test))
auc = roc_auc_score(y_test, pipe_mlp.predict_proba(X_test)[:,1])
print(f"MLP (con scaler) — F1={f1:.4f} AUC={auc:.4f}")
# Sin escalamiento (incorrecto, para demostrar el efecto)
pipe_mlp_raw = Pipeline([
("clf", MLPClassifier(
hidden_layer_sizes=(64, 32),
max_iter=300, random_state=SEED
))
])
pipe_mlp_raw.fit(X_train, y_train)
f1_raw = f1_score(y_test, pipe_mlp_raw.predict(X_test))
auc_raw = roc_auc_score(y_test, pipe_mlp_raw.predict_proba(X_test)[:,1])
print(f"MLP (sin scaler) — F1={f1_raw:.4f} AUC={auc_raw:.4f}")
```
---
# Recordatorio de métricas
Las métricas de clasificación se derivan de la matriz de confusión, que organiza las predicciones en cuatro categorías:
| | Predicho Positivo | Predicho Negativo |
|---|:---:|:---:|
| **Real Positivo** | TP | FN |
| **Real Negativo** | FP | TN |
Las métricas principales son:
$$\text{Accuracy} = \frac{TP + TN}{TP + TN + FP + FN}$$
$$\text{Precision} = \frac{TP}{TP + FP}$$
$$\text{Recall (Sensibilidad)} = \frac{TP}{TP + FN}$$
$$F_1 = \frac{2 \cdot \text{Precision} \cdot \text{Recall}}{\text{Precision} + \text{Recall}}$$
[@powers2011evaluation; @pedregosa2011sklearn]
## Curva ROC y AUC
La curva ROC (*Receiver Operating Characteristic*) [@fawcett2006roc] representa la tasa de verdaderos positivos (recall) en función de la tasa de falsos positivos para todos los umbrales de decisión posibles:
$$\text{TPR} = \frac{TP}{TP + FN} \qquad \text{FPR} = \frac{FP}{FP + TN}$$
El área bajo la curva ROC (AUC, *Area Under the Curve*) resume el rendimiento del clasificador en todos los thresholds. Un clasificador perfecto tiene AUC = 1.0; un clasificador aleatorio tiene AUC = 0.5.
## Curva Precision-Recall y Average Precision
La curva Precision-Recall [@saito2015precision] representa precision en función de recall para todos los thresholds. Es más informativa que la curva ROC cuando la clase positiva es rara, ya que la curva ROC puede ser engañosamente optimista en presencia de muchos verdaderos negativos.
La métrica *Average Precision* (AP) es el área bajo la curva Precision-Recall, calculada como:
$$\text{AP} = \sum_k (R_k - R_{k-1}) \cdot P_k$$
donde $P_k$ y $R_k$ son la precisión y el recall para el threshold $k$.
## Selección de métricas según el contexto
La elección de la métrica de evaluación depende del contexto del problema:
- **Clases balanceadas:** accuracy puede ser una métrica razonable
- **Clases desbalanceadas:** F1-score, ROC-AUC y AP son más informativas
- **Falsos negativos costosos** (p.ej., diagnóstico de enfermedades): maximizar recall
- **Falsos positivos costosos** (p.ej., spam, fraude con revisión manual costosa): maximizar precision
- **Balance entre ambos:** F1-score o F-beta con $\beta$ ajustado al costo relativo
- **Análisis del comportamiento para todos los thresholds:** curvas ROC o Precision-Recall
---
# Ajuste de thresholds de decisión
## Motivación
Por defecto, `predict()` clasifica una observación como positiva si su probabilidad estimada supera 0.5. Sin embargo, este umbral es arbitrario y no necesariamente óptimo para el objetivo del problema. En diagnóstico médico, donde un falso negativo puede tener consecuencias graves, puede convenir bajar el threshold para aumentar el recall a costa de reducir la precision.
## predict_proba y decision_function
Los clasificadores de scikit-learn exponen las probabilidades estimadas mediante `predict_proba()`. Para algunos modelos (como SVM), también está disponible `decision_function()`, que produce un *score* continuo cuyo signo determina la clase predicha.
```{python}
#| label: proba-capitulo
pipe_rf.fit(X_train, y_train)
probs = pipe_rf.predict_proba(X_test)
print("Probabilidades para las primeras 5 observaciones:")
print(pd.DataFrame(probs, columns=["P(maligno=0)", "P(benigno=1)"]).head())
```
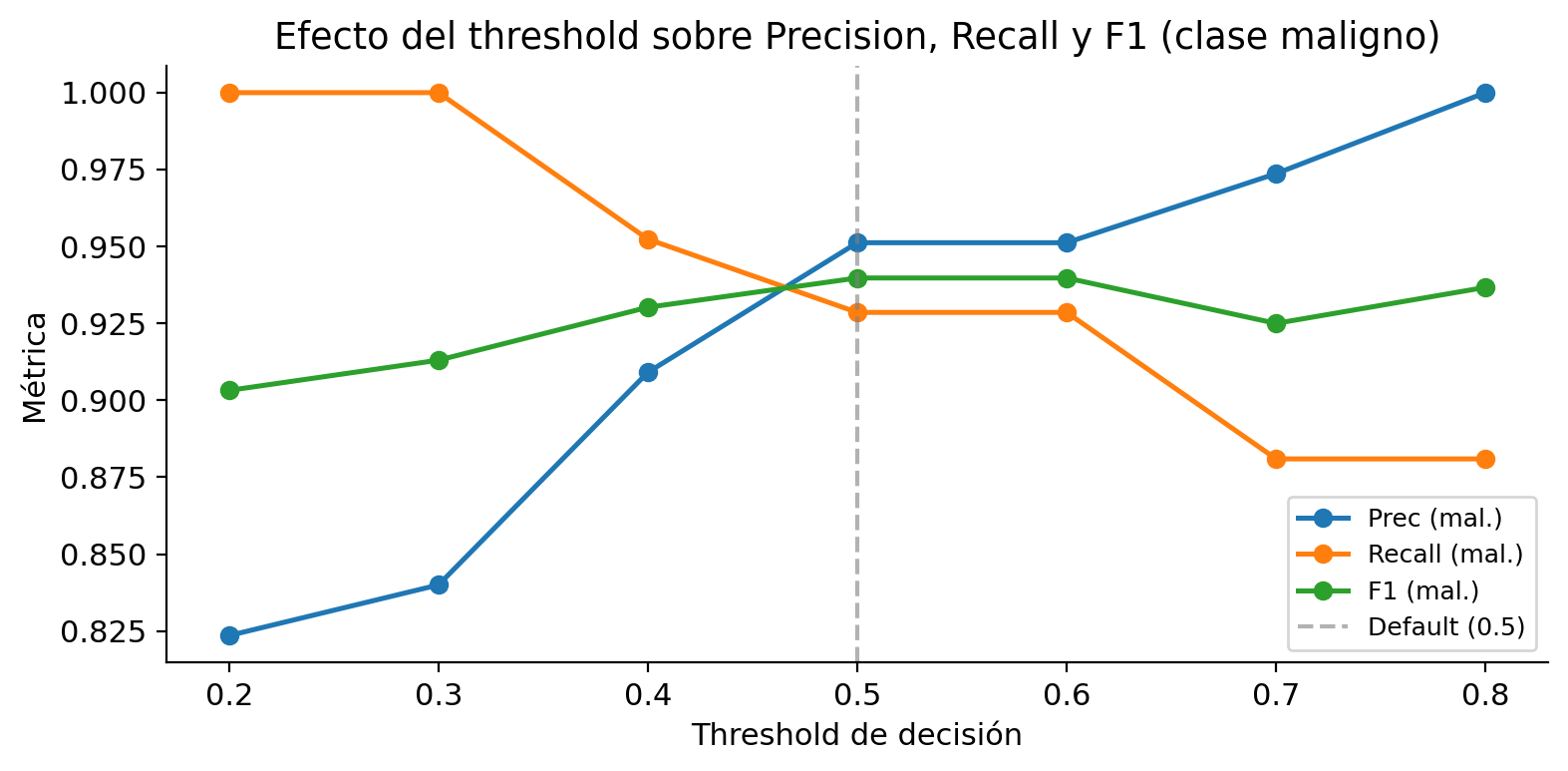
## Comparación de thresholds
Para clasificar una observación como maligna usamos $P(\text{maligno}) \geq \theta$, donde $\theta$ es el threshold. Al reducir $\theta$, clasificamos más observaciones como malignas, lo que aumenta el recall pero reduce la precision.
```{python}
#| label: thresholds-capitulo
from sklearn.metrics import precision_score, recall_score, f1_score
probs_mal = pipe_rf.predict_proba(X_test)[:, 0]
thresholds = [0.20, 0.30, 0.40, 0.50, 0.60, 0.70, 0.80]
filas = []
for th in thresholds:
y_pred = np.where(probs_mal >= th, 0, 1)
filas.append({
"Threshold": th,
"Prec (mal.)": precision_score(y_test, y_pred, pos_label=0, zero_division=0),
"Recall (mal.)":recall_score(y_test, y_pred, pos_label=0, zero_division=0),
"F1 (mal.)": f1_score(y_test, y_pred, pos_label=0, zero_division=0),
"FN": ((y_pred == 1) & (y_test.values == 0)).sum(),
"FP": ((y_pred == 0) & (y_test.values == 1)).sum(),
})
df_th = pd.DataFrame(filas).set_index("Threshold")
print(df_th.round(4).to_string())
```
```{python}
#| label: viz-thresholds-capitulo
fig, ax = plt.subplots(figsize=(8, 4))
df_th[["Prec (mal.)", "Recall (mal.)", "F1 (mal.)"]].plot(ax=ax, marker="o", linewidth=1.8)
ax.axvline(0.5, color="gray", linestyle="--", alpha=0.6, label="Default (0.5)")
ax.set_xlabel("Threshold de decisión")
ax.set_ylabel("Métrica")
ax.set_title("Efecto del threshold sobre Precision, Recall y F1 (clase maligno)")
ax.legend(fontsize=9)
plt.tight_layout()
plt.show()
```
## Consideraciones prácticas sobre el threshold
- El threshold óptimo depende del **costo relativo** de FP y FN, no de las métricas en el test set
- El threshold debe elegirse sobre un **conjunto de validación**, no sobre el test set
- La curva ROC y la curva Precision-Recall son herramientas para analizar el comportamiento del clasificador en **todos** los thresholds posibles antes de comprometerse con uno
---
# Comparación entre modelos
La comparación válida entre modelos requiere que todos sean evaluados sobre el **mismo conjunto de prueba**, con el **mismo preprocesamiento** y el **mismo criterio de evaluación**.
```{python}
#| label: comparacion-capitulo
from sklearn.ensemble import GradientBoostingClassifier, HistGradientBoostingClassifier
from sklearn.metrics import accuracy_score
modelos_todos = {
"Logistic Regression": Pipeline([
("scaler", StandardScaler()),
("clf", LogisticRegression(max_iter=1000, random_state=SEED))
]),
"CART (Gini)": Pipeline([
("clf", DecisionTreeClassifier(criterion="gini", max_depth=5, random_state=SEED))
]),
"ID3-proxy (Entropy)": Pipeline([
("clf", DecisionTreeClassifier(criterion="entropy", max_depth=5, random_state=SEED))
]),
"Random Forest": Pipeline([
("clf", RandomForestClassifier(n_estimators=100, random_state=SEED))
]),
"Gradient Boosting": Pipeline([
("clf", GradientBoostingClassifier(
n_estimators=100, learning_rate=0.1, max_depth=3, random_state=SEED
))
]),
"MLP": Pipeline([
("scaler", StandardScaler()),
("clf", MLPClassifier(hidden_layer_sizes=(64, 32), max_iter=300, random_state=SEED))
]),
}
filas_comp = []
for nombre, pipe in modelos_todos.items():
pipe.fit(X_train, y_train)
y_pred = pipe.predict(X_test)
y_prob = pipe.predict_proba(X_test)[:, 1]
filas_comp.append({
"Modelo": nombre,
"Accuracy": accuracy_score(y_test, y_pred),
"F1": f1_score(y_test, y_pred),
"ROC-AUC": roc_auc_score(y_test, y_prob),
"Recall": recall_score(y_test, y_pred),
"Precision": precision_score(y_test, y_pred),
})
df_comp = pd.DataFrame(filas_comp).set_index("Modelo")
print(df_comp.sort_values("ROC-AUC", ascending=False).round(4).to_string())
```
---
# Buenas prácticas para evaluación
Las siguientes prácticas garantizan que los resultados del experimento sean válidos, reproducibles e interpretables:
1. **Usar pipelines** que encapsulen preprocesamiento y modelo, evitando leakage
2. **Fijar semillas aleatorias** con `random_state` en todos los modelos y la partición
3. **Usar `stratify`** al dividir el dataset cuando las clases están desbalanceadas
4. **Comparar modelos con la misma partición** y el mismo preprocesamiento
5. **Reportar múltiples métricas**, eligiendo las más relevantes para el contexto del problema
6. **Usar validación cruzada** para seleccionar hiperparámetros, no el test set
7. **No usar el test set repetidamente** para ajustar decisiones de diseño
8. **Analizar la matriz de confusión** junto con las métricas agregadas
9. **Verificar el balance de clases** antes de elegir las métricas de evaluación
10. **Documentar** todos los parámetros, versiones de librerías y semillas usadas
---
# Errores comunes en experimentos de clasificación
A continuación se describen los errores más frecuentes al diseñar y ejecutar experimentos de clasificación:
**Evaluación en datos de entrenamiento.** Evaluar el modelo sobre los mismos datos con los que fue entrenado produce resultados artificialmente optimistas que no reflejan la capacidad de generalización.
**Data leakage por escalamiento.** Ajustar el scaler sobre todo el dataset (incluyendo test) antes del split permite que información del test set "contamine" el entrenamiento. La solución es usar pipelines.
**Evaluar solo con accuracy.** En datasets con desbalance de clases, un modelo que siempre predice la clase mayoritaria puede tener una accuracy alta pero un rendimiento nulo en la clase minoritaria. Métricas como F1-score, ROC-AUC y AP son más informativas en estos casos.
**No revisar el balance de clases.** Ignorar el desbalance de clases puede llevar a elegir métricas incorrectas y a modelos con rendimiento deficiente en la clase minoritaria.
**Comparar modelos con particiones distintas.** Si dos modelos se evalúan sobre particiones distintas, las diferencias observadas pueden deberse al azar, no a diferencias reales de rendimiento.
**No usar `random_state`.** Los resultados no son reproducibles y no pueden ser comparados entre ejecuciones.
**Redes neuronales sin escalamiento.** Las redes neuronales son sensibles a la escala de las variables de entrada. Sin escalamiento, el optimizador puede converger lentamente o a un mínimo deficiente.
**Ajustar hiperparámetros mirando el test set.** Si se usa el test set para elegir hiperparámetros, el modelo está implícitamente siendo ajustado sobre el test, lo que produce una evaluación optimista.
**Interpretar AUC sin contexto.** Un AUC alto no garantiza que el modelo sea útil para el problema si el threshold elegido produce demasiados falsos negativos o falsos positivos.
---
# Retos prácticos del flujo de clasificación
Más allá de los errores técnicos, existen retos conceptuales y de diseño que aparecen en cualquier proyecto de clasificación real:
**Identificación de la clase positiva.** La elección de qué clase se considera "positiva" afecta directamente el cálculo de precision, recall y F1. Esta decisión debe tomarse en función del objetivo del problema.
**Costo asimétrico de los errores.** En muchos problemas reales, un falso negativo no es equivalente a un falso positivo. El diseño del experimento debe reflejar estas asimetrías en la elección de métricas y thresholds.
**Escalas heterogéneas.** Variables con escalas muy distintas afectan a modelos sensibles como regresión logística, KNN y redes neuronales. Los árboles de decisión y Random Forest son invariantes al escalamiento.
**Variables categóricas y numéricas mixtas.** Cuando el dataset contiene ambos tipos de variables, es necesario usar `ColumnTransformer` para aplicar transformaciones distintas a cada grupo.
**Reproducibilidad del experimento.** Un experimento reproducible requiere fijar semillas, documentar versiones de librerías y guardar el estado del pipeline entrenado.
**Selección del threshold en producción.** El threshold óptimo para producción debe decidirse con datos de validación, considerando el costo relativo de FP y FN en el contexto del negocio o la aplicación.
---
# Conclusión
Este capítulo ha mostrado que diseñar un experimento de clasificación correcto es más complejo que simplemente llamar `.fit()`. Las decisiones sobre la partición de datos, el preprocesamiento, la elección de métricas, la comparación entre modelos y el ajuste de thresholds tienen un impacto directo sobre la validez y la utilidad de los resultados.
Los modelos estudiados — regresión logística, CART, ID3, Random Forest, Gradient Boosting y MLP — representan distintos sesgos inductivos y compromisos entre interpretabilidad, flexibilidad y rendimiento. Ninguno es universalmente superior; la elección del modelo depende del dataset, el objetivo del problema y los recursos disponibles.
La práctica rigurosa de clasificación supervisada combina conocimiento algorítmico, diseño experimental cuidadoso e interpretación crítica de los resultados.
---
# Referencias {.unnumbered}
::: {#refs}
:::