---
title: "Árboles de regresión lineal, ensambles y vecinos cercanos"
author: "Diego Villalba"
date: today
lang: es
format:
html:
toc: true
toc-depth: 3
toc-title: "Contenido"
number-sections: true
code-fold: true
code-tools: true
code-summary: "Mostrar código"
fig-align: center
theme: cosmo
highlight-style: github
smooth-scroll: true
pdf:
toc: true
toc-depth: 3
number-sections: true
documentclass: scrbook
papersize: letter
fontsize: 11pt
geometry: margin=2.5cm
keep-tex: false
execute:
echo: true
warning: false
message: false
cache: false
bibliography: referencias_arboles_regresion_lineal.bib
crossref:
fig-title: "Figura"
tbl-title: "Tabla"
eq-prefix: "Ec."
---
Los árboles de decisión constituyen una familia de métodos de aprendizaje supervisado basada en reglas. A diferencia de un modelo lineal global, que intenta describir todo el conjunto de datos mediante una sola ecuación, un árbol divide el espacio de variables en regiones más pequeñas y ajusta una predicción simple en cada una. Esta idea es especialmente útil cuando la relación entre las variables explicativas y la variable respuesta no es lineal, cambia por zonas o involucra interacciones difíciles de especificar manualmente [@breiman1984classification; @hastie2009elements].
En problemas de regresión, el objetivo es predecir una variable numérica. Por ejemplo, podríamos estimar el precio de una laptop a partir de su memoria RAM, el tamaño de pantalla, el procesador o la marca; o bien estimar el precio de una vivienda con base en su área, ubicación y antigüedad. Un árbol de regresión construye reglas del tipo “si una variable cae por debajo de cierto umbral, sigue una rama; en caso contrario, sigue otra”. Al final de cada rama se encuentra una hoja, donde la predicción suele ser el promedio de las observaciones de entrenamiento que llegaron a esa región.
Este capítulo desarrolla la lógica de los árboles de regresión, su interpretación como funciones por partes, el criterio de error usado para elegir divisiones, la poda para controlar el sobreajuste y las métricas básicas de evaluación. Posteriormente se introducen métodos de ensamble basados en árboles, en particular bagging, random forests y boosting. Finalmente se presenta el algoritmo de vecinos más cercanos como contraste con los árboles: un método supervisado basado en distancia y aprendizaje perezoso.
## De la regresión lineal a modelos por regiones
La regresión lineal múltiple modela una respuesta continua $y$ como combinación lineal de variables explicativas $x_1,\ldots,x_p$:
$$
y_i = \beta_0 + \beta_1 x_{i1}+\cdots+\beta_p x_{ip}+\varepsilon_i.
$$
Su principal ventaja es la interpretabilidad de los coeficientes: cada $\beta_j$ mide el cambio esperado en $y$ ante un incremento unitario de $x_j$, manteniendo constantes las demás variables. Sin embargo, esta estructura impone una forma funcional global. Cuando la relación entre $X$ e $y$ cambia en distintas regiones del espacio, una sola ecuación puede resultar demasiado rígida.
Los árboles de regresión responden a esta dificultad mediante particionamiento recursivo. En lugar de buscar una única superficie global, dividen el espacio de predictores en regiones y asignan una predicción constante a cada región. Esto produce un modelo no paramétrico, flexible e interpretable [@james2021introduction].
::: {.callout-note appearance=“minimal”}
Definición. Un árbol de regresión es un modelo que particiona el espacio de predictores en regiones disjuntas $R_1,\ldots,R_J$ y predice un valor constante $c_h$ dentro de cada región:
$$
\hat f(x)=\sum_{h=1}^{J} c_h\,\mathbb{I}(x\in R_h).
$$ {#eq-tree-piecewise}
:::
En esta expresión, $x$ representa una nueva observación; $R_h$ es una región del espacio de variables; $c_h$ es el valor asignado a esa región, usualmente el promedio de las respuestas observadas dentro de ella; y $\mathbb{I}(x\in R_h)$ es una función indicadora que vale uno si $x$ pertenece a la región $R_h$ y cero en caso contrario.
::: {.callout-note appearance=“minimal”}
Definición. La función indicadora de una región $R_h$ se define como:
$$
\mathbb{I}(x\in R_h)=
\begin{cases}
1, & \text{si } x\in R_h,\\
0, & \text{si } x\notin R_h.
\end{cases}
$$ {#eq-indicator}
:::
## Clasificadores no lineales y modelos por partes
En problemas de clasificación, un clasificador lineal separa clases mediante hiperplanos. Si los datos son linealmente separables, esta estrategia puede ser suficiente. Sin embargo, en muchos problemas reales las fronteras entre clases son curvas, fragmentadas o dependen de interacciones entre variables. En estos casos existen dos alternativas: mantener un clasificador lineal y aceptar ciertos errores, o usar clasificadores no lineales capaces de aproximar fronteras más flexibles.
Una forma pedagógica de construir no linealidad es usar piezas lineales. La idea es dividir una clase en subclases o regiones locales y asignar a cada subregión un discriminante lineal. La frontera global resultante deja de ser un solo hiperplano y se convierte en una unión de segmentos o caras lineales. Esta lógica anticipa dos ideas que aparecen más adelante: los árboles, que dividen el espacio mediante cortes recursivos, y los vecinos cercanos, que llevan la idea local al extremo de tratar cada muestra como una referencia de decisión [@duda2001pattern; @hastie2009elements].
::: {.callout-note appearance=“minimal”}
Definición. Un clasificador lineal por partes asigna a cada clase $\omega_i$ un conjunto de subclases $\omega_i^1,\ldots,\omega_i^{\ell_i}$, cada una con un discriminante lineal $g_i^\ell(x)$, y define el discriminante de la clase como el máximo local:
$$
g_i(x)=\max_{\ell=1,\ldots,\ell_i} g_i^\ell(x),\qquad
\hat y(x)=\arg\max_{i=1,\ldots,c} g_i(x).
$$ {#eq-piecewise-linear-discriminant}
:::
Si se escribe $g_i^\ell(x)=w_{i\ell}^{\top}x+w_{i\ell,0}$, cada subclase aporta una frontera lineal. El operador máximo selecciona la subregión de la clase que mejor explica el punto $x$. Cuando el número de subclases aumenta, la frontera puede aproximar formas no lineales cada vez más complejas. No obstante, esta flexibilidad tiene un costo: se deben decidir cuántas subclases usar, cómo inicializarlas y cómo evitar que el modelo memorice ruido.
Desde esta perspectiva, los árboles de decisión construyen una aproximación por partes, pero con piezas constantes en regresión y fronteras alineadas con los ejes. KNN, por su parte, puede verse como un caso extremadamente local: en lugar de aprender subclases explícitas, conserva los ejemplos de entrenamiento y decide usando distancias.
## Funciones por escalones
Un árbol de regresión produce una función por escalones. Para una sola variable $x$, un modelo simple podría definirse como:
$$
\hat f(x)=
\begin{cases}
100, & x<10,\\
200, & 10\le x<20,\\
300, & x\ge 20.
\end{cases}
$$
Este modelo no intenta ajustar una recta. En su lugar, divide el eje de $x$ en intervalos y asigna una predicción constante a cada intervalo. En dos variables, las regiones suelen ser rectángulos; en $p$ variables, se interpretan como hiperrectángulos. Por ejemplo, con edad, salario y experiencia, una regla podría ser:
$$
\text{edad}<30 \quad \text{y} \quad \text{salario}>5000.
$$
Este tipo de reglas es fácil de leer, lo que explica la popularidad de los árboles en contextos donde la interpretación es importante. No obstante, también implica una limitación geométrica: las fronteras de decisión son paralelas a los ejes, por lo que algunas relaciones oblicuas o altamente correlacionadas pueden necesitar muchos cortes para aproximarse bien [@hastie2009elements].
## Predicción dentro de una región
Una vez que el árbol define una región $R_h$, la predicción óptima bajo pérdida cuadrática es el promedio de los valores reales dentro de esa región. Supongamos que en una región hay observaciones con respuestas $y_1,\ldots,y_{n_h}$. La predicción constante $c_h$ se elige minimizando la suma de errores cuadrados:
$$
\sum_{i:x_i\in R_h}(y_i-c_h)^2.
$$
La solución es el promedio muestral.
::: {.callout-note appearance=“minimal”}
Definición. La predicción de una hoja $R_h$ en un árbol de regresión, bajo error cuadrático, es el promedio de las respuestas que caen en esa hoja:
$$
\hat c_h = \frac{1}{|R_h|}\sum_{i:x_i\in R_h} y_i.
$$ {#eq-leaf-mean}
:::
Por ejemplo, si una región contiene los valores reales $20,40,100,120$, su predicción antes de dividir es:
$$
\hat c=\frac{20+40+100+120}{4}=70.
$$
Si esa región se divide en dos grupos, $R_1=\{20,40\}$ y $R_2=\{100,120\}$, las nuevas predicciones son:
$$
\hat c_1=30,\qquad \hat c_2=110.
$$
La división es útil porque cada grupo queda descrito por un promedio más representativo de sus valores.
## Error total y criterio de división
El árbol decide sus cortes evaluando cuánto disminuye el error al dividir una región. Para regresión, el criterio más común es la suma de errores cuadrados dentro de las hojas, también conocida como residual sum of squares (RSS) [@breiman1984classification; @james2021introduction].
::: {.callout-note appearance=“minimal”}
Definición. El error cuadrático total de un árbol con regiones $R_1,\ldots,R_J$ es:
$$
D(T)=\sum_{h=1}^{J}\sum_{i:x_i\in R_h}(y_i-\hat c_h)^2.
$$ {#eq-tree-error}
:::
Cuando se propone dividir una región $R$ en dos subregiones $R_L$ y $R_R$, el árbol compara el error antes y después de la división. El error después de dividir es:
$$
D_{\text{split}} = \sum_{i:x_i\in R_L}(y_i-\hat c_L)^2+
\sum_{i:x_i\in R_R}(y_i-\hat c_R)^2.
$$
::: {.callout-note appearance=“minimal”}
Definición. La ganancia de una división se define como la reducción de error producida por el corte:
$$
\text{Gain}=D_{\text{antes}}-D_{\text{después}}.
$$ {#eq-gain}
:::
Una división es buena si reduce mucho el error. Si la reducción es pequeña, el corte puede no justificar el aumento de complejidad. Durante el entrenamiento, el algoritmo prueba muchas divisiones candidatas: por ejemplo, cortes en edad, salario o experiencia; y dentro de cada variable, varios umbrales posibles. Se elige la división que produce la mayor reducción de error.
```{=html}
<div id="rss-wrap" style="font-family:Georgia,serif;max-width:700px;margin:2em auto;background:#f9fafc;border:1px solid #d0d7e3;border-radius:10px;padding:20px 24px;box-shadow:0 2px 12px rgba(30,60,120,.07);">
<div style="text-align:center;margin-bottom:12px;">
<span style="font-size:1.05em;font-weight:bold;color:#1a2e45;">Criterio de división RSS — explorador interactivo</span><br>
<span style="font-size:0.81em;color:#666;">Arrastra el umbral de corte y observa cómo cambia el RSS antes y después de la partición</span>
</div>
<div style="display:flex;align-items:center;gap:12px;margin-bottom:10px;flex-wrap:wrap;">
<label style="font-size:0.83em;color:#444;">Umbral RAM = <strong id="rss-tv">6.0</strong> GB</label>
<input id="rss-sl" type="range" min="4.5" max="15.5" step="0.5" value="6"
style="flex:1;min-width:150px;accent-color:#1a3a5c;">
</div>
<div id="rss-info" style="text-align:center;font-size:0.82em;padding:5px 10px;background:#fff;border:1px solid #e0e5ed;border-radius:5px;margin-bottom:10px;min-height:22px;"></div>
<canvas id="rss-cv" style="display:block;width:100%;border-radius:6px;border:1px solid #e0e5ed;background:#fff;"></canvas>
</div>
<script>
(function(){
const RAM=[4,4,8,8,16,16], Y=[6500,7200,10500,12000,21000,24000], N=6;
const cv=document.getElementById('rss-cv');
const W=680,H=260,DPR=window.devicePixelRatio||1;
cv.width=W*DPR;cv.height=H*DPR;cv.style.width=W+'px';cv.style.height=H+'px';
const ctx=cv.getContext('2d');ctx.scale(DPR,DPR);
const LP={l:52,r:W/2-8,t:20,b:220},RP={l:W/2+8,r:W-10,t:20,b:220};
const LW=LP.r-LP.l,LH=LP.b-LP.t,RW=RP.r-RP.l,RH=RP.b-RP.t;
const xMn=2,xMx=18,yMn=0,yMx=26000;
const sx=x=>LP.l+(x-xMn)/(xMx-xMn)*LW;
const sy=y=>LP.t+LH-(y-yMn)/(yMx-yMn)*LH;
function rss(arr){const m=arr.reduce((s,v)=>s+v,0)/arr.length;return arr.reduce((s,v)=>s+(v-m)**2,0);}
function draw(t){
ctx.clearRect(0,0,W,H);
const yL=Y.filter((_,i)=>RAM[i]<t),yR=Y.filter((_,i)=>RAM[i]>=t);
const rssTotal=rss(Y),rssSplit=(yL.length?rss(yL):0)+(yR.length?rss(yR):0);
const gain=rssTotal-rssSplit;
const meanAll=Y.reduce((s,v)=>s+v,0)/N;
const meanL=yL.length?yL.reduce((s,v)=>s+v,0)/yL.length:null;
const meanR=yR.length?yR.reduce((s,v)=>s+v,0)/yR.length:null;
ctx.fillStyle='#f8f9fb';ctx.fillRect(LP.l,LP.t,LW,LH);
ctx.strokeStyle='#e8ecf3';ctx.lineWidth=0.8;
[4,8,12,16].forEach(x=>{ctx.beginPath();ctx.moveTo(sx(x),LP.t);ctx.lineTo(sx(x),LP.b);ctx.stroke();});
[0,5000,10000,15000,20000,25000].forEach(y=>{ctx.beginPath();ctx.moveTo(LP.l,sy(y));ctx.lineTo(LP.r,sy(y));ctx.stroke();});
ctx.strokeStyle='#9aa3b5';ctx.lineWidth=1.2;
ctx.beginPath();ctx.moveTo(LP.l,LP.t);ctx.lineTo(LP.l,LP.b);ctx.lineTo(LP.r,LP.b);ctx.stroke();
ctx.fillStyle='#888';ctx.font='10px Georgia,serif';ctx.textAlign='center';
[4,8,12,16].forEach(x=>ctx.fillText(x+'GB',sx(x),LP.b+13));
ctx.textAlign='right';
[0,10000,20000].forEach(y=>ctx.fillText((y/1000).toFixed(0)+'k',LP.l-4,sy(y)+3));
ctx.fillStyle='#1a2e45';ctx.font='bold 11px Georgia,serif';ctx.textAlign='center';
ctx.fillText('Datos y predicción del árbol',LP.l+LW/2,LP.t-5);
ctx.strokeStyle='rgba(100,100,100,0.3)';ctx.lineWidth=1;ctx.setLineDash([3,3]);
ctx.beginPath();ctx.moveTo(sx(xMn),sy(meanAll));ctx.lineTo(sx(xMx),sy(meanAll));ctx.stroke();
ctx.setLineDash([]);
if(meanL!==null){
ctx.strokeStyle='rgba(37,99,235,0.6)';ctx.lineWidth=2;ctx.setLineDash([5,3]);
ctx.beginPath();ctx.moveTo(sx(xMn),sy(meanL));ctx.lineTo(sx(t),sy(meanL));ctx.stroke();
ctx.fillStyle='#2563eb';ctx.font='bold 10px Georgia,serif';ctx.textAlign='center';
ctx.fillText('ĉ₁='+meanL.toFixed(0),sx((xMn+t)/2),sy(meanL)-7);
}
if(meanR!==null){
ctx.strokeStyle='rgba(220,38,38,0.6)';ctx.lineWidth=2;ctx.setLineDash([5,3]);
ctx.beginPath();ctx.moveTo(sx(t),sy(meanR));ctx.lineTo(sx(xMx),sy(meanR));ctx.stroke();
ctx.fillStyle='#dc2626';ctx.font='bold 10px Georgia,serif';ctx.textAlign='center';
ctx.fillText('ĉ₂='+meanR.toFixed(0),sx((t+xMx)/2),sy(meanR)-7);
}
ctx.setLineDash([]);
if(t>xMn&&t<xMx){
ctx.strokeStyle='#f59e0b';ctx.lineWidth=2;
ctx.beginPath();ctx.moveTo(sx(t),LP.t);ctx.lineTo(sx(t),LP.b);ctx.stroke();
ctx.fillStyle='#f59e0b';ctx.font='bold 10px Georgia,serif';ctx.textAlign='left';
ctx.fillText('t='+t,sx(t)+4,LP.t+12);
}
RAM.forEach((r,i)=>{
const inL=r<t;
ctx.beginPath();ctx.arc(sx(r)+(i%2===0?-5:5),sy(Y[i]),6,0,2*Math.PI);
ctx.fillStyle=inL?'#2563ebcc':'#dc2626cc';ctx.fill();
ctx.strokeStyle=inL?'#1d4ed8':'#b91c1c';ctx.lineWidth=1.5;ctx.stroke();
});
ctx.fillStyle='#f8f9fb';ctx.fillRect(RP.l,RP.t,RW,RH);
ctx.fillStyle='#1a2e45';ctx.font='bold 11px Georgia,serif';ctx.textAlign='center';
ctx.fillText('Comparación RSS',RP.l+RW/2,RP.t-5);
const bars=[{lbl:'RSS sin\ndividir',val:rssTotal,col:'#9ca3af'},{lbl:'RSS con\ndivisión',val:rssSplit,col:gain>0?'#16a34a':'#dc2626'}];
const maxRSS=rssTotal*1.1,bW=RW*0.22;
bars.forEach((b,i)=>{
const bx=RP.l+RW*(i+1)/(bars.length+1)-bW/2;
const bh=(b.val/maxRSS)*(RH-40),by=RP.b-bh-4;
ctx.fillStyle=b.col+'bb';ctx.fillRect(bx,by,bW,bh);
ctx.strokeStyle=b.col;ctx.lineWidth=1.5;ctx.strokeRect(bx,by,bW,bh);
ctx.fillStyle='#1a2e45';ctx.font='bold 10px Georgia,serif';ctx.textAlign='center';
const v=b.val>=1e6?(b.val/1e6).toFixed(2)+'M':(b.val/1e3).toFixed(0)+'k';
ctx.fillText(v,bx+bW/2,by-5);
ctx.fillStyle=b.col;ctx.font='10px Georgia,serif';
b.lbl.split('\n').forEach((l,j)=>ctx.fillText(l,bx+bW/2,RP.b+13+j*12));
});
const gainPct=(gain/rssTotal*100).toFixed(1);
const gc=gain>rssTotal*0.3?'#16a34a':gain>0?'#ca8a04':'#dc2626';
ctx.fillStyle=gc;ctx.font='bold 11px Georgia,serif';ctx.textAlign='center';
ctx.fillText('Ganancia: '+gainPct+'%',RP.l+RW/2,RP.t+RH*0.5);
document.getElementById('rss-info').innerHTML=
`RSS sin dividir = <strong>${(rssTotal/1e6).toFixed(3)}M</strong> · `+
`RSS con división = <strong>${(rssSplit/1e6).toFixed(3)}M</strong> · `+
`Ganancia = <strong style="color:${gc}">${gainPct}%</strong>`+
(yL.length===0||yR.length===0?'  <em style="color:#aaa">(sin datos en una región)</em>':'');
}
document.getElementById('rss-sl').addEventListener('input',function(){
const v=parseFloat(this.value);
document.getElementById('rss-tv').textContent=v.toFixed(1);
draw(v);
});
draw(6);
})();
</script>
```
## Particionamiento recursivo
El entrenamiento de un árbol se realiza de forma codiciosa. Primero se busca el mejor corte para todo el conjunto de entrenamiento. Luego, dentro de cada región resultante, se vuelve a buscar el mejor corte local. Este procedimiento se repite hasta que se cumple un criterio de parada: profundidad máxima, número mínimo de observaciones por hoja, reducción mínima de error o tamaño mínimo de nodo.
El algoritmo básico puede resumirse así:
1. Comenzar con todos los datos en una sola región.
2. Para cada variable y cada punto de corte candidato, calcular el error después de dividir.
3. Elegir la división que más reduzca el error.
4. Repetir el proceso dentro de cada subregión.
5. Detener el crecimiento del árbol según reglas de control de complejidad.
El enfoque es eficiente y produce reglas interpretables, pero no garantiza encontrar el árbol globalmente óptimo. La búsqueda exacta del mejor árbol es combinatoriamente costosa, por lo que los algoritmos prácticos usan heurísticas codiciosas [@breiman1984classification].
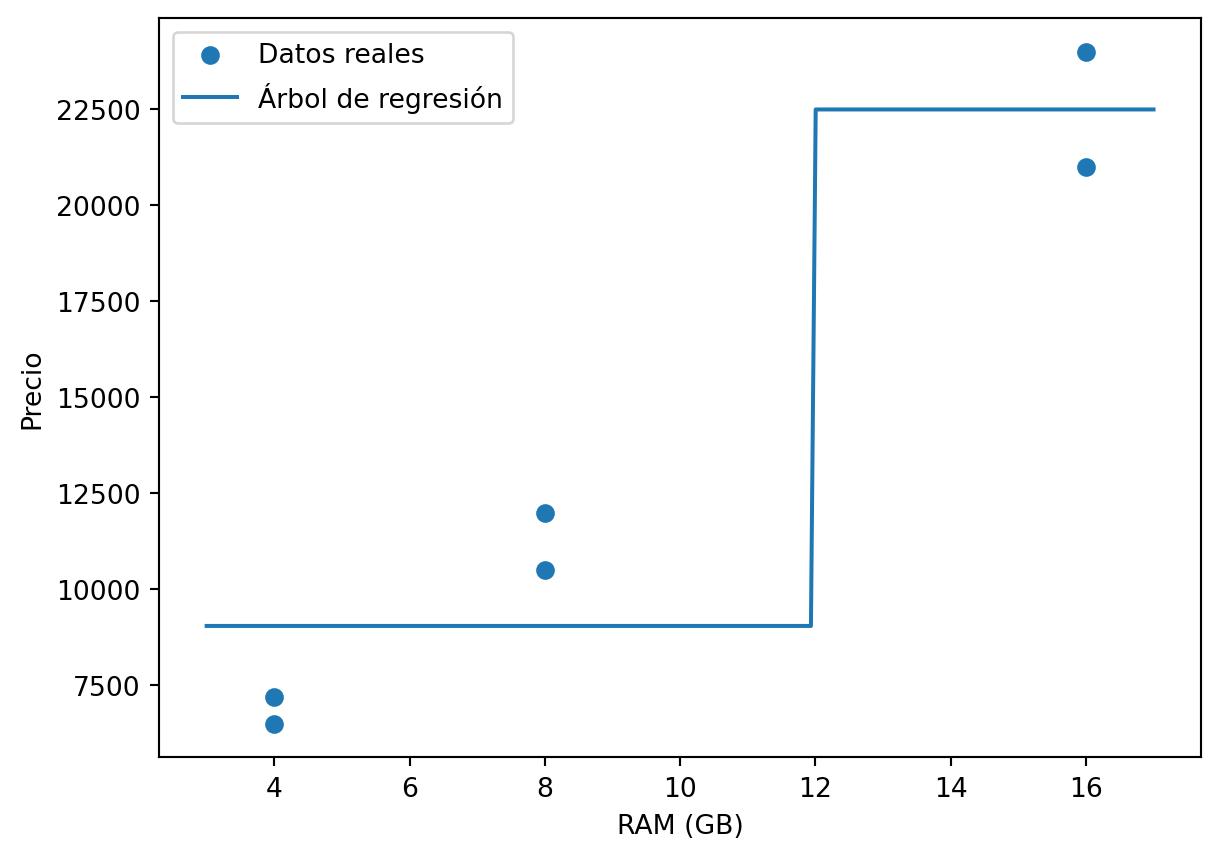
## Ejemplo manual: precio de laptops según RAM
Consideremos un conjunto de datos pequeño donde se desea predecir el precio de una laptop a partir de la memoria RAM.
| Laptop | RAM | Precio real |
|---|---:|---:|
| A | 4 GB | 6500 |
| B | 4 GB | 7200 |
| C | 8 GB | 10500 |
| D | 8 GB | 12000 |
| E | 16 GB | 21000 |
| F | 16 GB | 24000 |
Si no se divide el árbol, la predicción para todas las laptops es el promedio global:
$$
\bar y=\frac{6500+7200+10500+12000+21000+24000}{6}=13533.33.
$$
El error sin dividir es:
$$
D_{\text{sin dividir}}=\sum_i(y_i-\bar y)^2=266{,}433{,}333.33.
$$
Ahora se propone el corte:
$$
\text{RAM}<12.
$$
Esto genera dos regiones:
$$
R_1=\{6500,7200,10500,12000\},\qquad R_2=\{21000,24000\}.
$$
Las predicciones de cada hoja son:
$$
\hat c_1=\frac{6500+7200+10500+12000}{4}=9050,
$$
$$
\hat c_2=\frac{21000+24000}{2}=22500.
$$
El error después de dividir es:
$$
D_{\text{con división}}=
(6500-9050)^2+(7200-9050)^2+(10500-9050)^2+(12000-9050)^2
+(21000-22500)^2+(24000-22500)^2.
$$
Al calcular:
$$
D_{\text{con división}}=25{,}230{,}000.
$$
La mejora relativa es:
$$
\frac{D_{\text{sin dividir}}-D_{\text{con división}}}{D_{\text{sin dividir}}}
=\frac{266{,}433{,}333.33-25{,}230{,}000}{266{,}433{,}333.33}
\approx 0.9053.
$$
Por tanto, el corte reduce el error aproximadamente en $90.53\%$. El árbol resultante tiene una regla clara: si la RAM es menor que 12 GB, predice 9050; si es mayor o igual que 12 GB, predice 22500.
## Implementación en Python
El siguiente ejemplo reproduce la idea anterior con `scikit-learn`. Aunque el conjunto de datos es muy pequeño, permite observar cómo un árbol de regresión aprende divisiones por umbral.
```{python}
import numpy as np
import pandas as pd
from sklearn.tree import DecisionTreeRegressor, export_text
from sklearn.metrics import mean_squared_error, r2_score
# Datos de ejemplo
X = pd.DataFrame({
"RAM_GB": [4, 4, 8, 8, 16, 16]
})
y = np.array([6500, 7200, 10500, 12000, 21000, 24000])
# Árbol pequeño de regresión
tree = DecisionTreeRegressor(max_depth=1, random_state=42)
tree.fit(X, y)
pred = tree.predict(X)
print(export_text(tree, feature_names=list(X.columns)))
print("Predicciones:", pred)
print("MSE:", mean_squared_error(y, pred))
print("R2:", r2_score(y, pred))
```
El árbol de profundidad uno aprende un único corte. En un problema real se usaría una división entrenamiento-prueba o validación cruzada para estimar el desempeño fuera de muestra.
```{python}
import matplotlib.pyplot as plt
ram_grid = np.linspace(3, 17, 200).reshape(-1, 1)
pred_grid = tree.predict(pd.DataFrame(ram_grid, columns=["RAM_GB"]))
plt.figure()
plt.scatter(X["RAM_GB"], y, label="Datos reales")
plt.plot(ram_grid, pred_grid, label="Árbol de regresión")
plt.xlabel("RAM (GB)")
plt.ylabel("Precio")
plt.legend()
plt.show()
```
## Sobreajuste y poda
Un árbol puede seguir dividiendo hasta que cada hoja contenga muy pocas observaciones. En el extremo, podría memorizar los datos de entrenamiento. Esto reduce el error dentro de la muestra, pero aumenta el riesgo de fallar con datos nuevos. Este fenómeno se conoce como sobreajuste.
Para evitarlo, se controla la complejidad del árbol. Una estrategia clásica es la poda por costo-complejidad, que penaliza el número de hojas del árbol [@breiman1984classification; @scikit-learn-tree].
::: {.callout-note appearance=“minimal”}
Definición. El criterio de costo-complejidad penaliza el error total del árbol más un término proporcional al número de hojas:
$$
C_{\alpha}(T)=\sum_{h=1}^{J}D_h+\alpha J.
$$ {#eq-cost-complexity}
:::
Aquí $D_h$ es el error de la hoja $h$, $J$ es el número de hojas y $\alpha\ge 0$ controla la penalización por complejidad. Si $\alpha$ es pequeño, se permite un árbol más grande; si $\alpha$ es grande, se castigan más las divisiones.
Consideremos dos árboles:
| Modelo | Error | Número de hojas $J$ | $\alpha$ | Costo total |
|---|---:|---:|---:|---:|
| Árbol pequeño | 500 | 3 | 20 | $500+20(3)=560$ |
| Árbol grande | 300 | 20 | 20 | $300+20(20)=700$ |
Aunque el árbol grande tiene menor error de entrenamiento, su costo total es mayor debido a la penalización por complejidad. Bajo este criterio, se preferiría el árbol pequeño.
En `scikit-learn`, este control puede implementarse mediante el parámetro `ccp_alpha`.
```{python}
from sklearn.model_selection import train_test_split
from sklearn.datasets import fetch_california_housing
from sklearn.metrics import mean_absolute_error
housing = fetch_california_housing(as_frame=True)
X = housing.data
y = housing.target
X_train, X_test, y_train, y_test = train_test_split(
X, y, test_size=0.25, random_state=42
)
for alpha in [0.0, 0.001, 0.01, 0.05]:
model = DecisionTreeRegressor(random_state=42, ccp_alpha=alpha)
model.fit(X_train, y_train)
y_pred = model.predict(X_test)
print({
"ccp_alpha": alpha,
"n_leaves": model.get_n_leaves(),
"MAE": mean_absolute_error(y_test, y_pred),
"R2": r2_score(y_test, y_pred)
})
```
```{=html}
<div id="td-wrap" style="font-family:Georgia,serif;max-width:700px;margin:2em auto;background:#f9fafc;border:1px solid #d0d7e3;border-radius:10px;padding:20px 24px;box-shadow:0 2px 12px rgba(30,60,120,.07);">
<div style="text-align:center;margin-bottom:12px;">
<span style="font-size:1.05em;font-weight:bold;color:#1a2e45;">Profundidad del árbol y sobreajuste</span><br>
<span style="font-size:0.81em;color:#666;">Aumenta la profundidad máxima y observa cómo el árbol memoriza los datos de entrenamiento</span>
</div>
<div style="display:flex;align-items:center;gap:12px;margin-bottom:10px;flex-wrap:wrap;">
<label style="font-size:0.83em;color:#444;">Profundidad máx = <strong id="td-dv">2</strong></label>
<input id="td-sl" type="range" min="1" max="7" step="1" value="2"
style="flex:1;min-width:150px;accent-color:#1a3a5c;">
</div>
<div id="td-info" style="text-align:center;font-size:0.82em;padding:5px 10px;background:#fff;border:1px solid #e0e5ed;border-radius:5px;margin-bottom:10px;min-height:22px;"></div>
<canvas id="td-cv" style="display:block;width:100%;border-radius:6px;border:1px solid #e0e5ed;background:#fff;"></canvas>
</div>
<script>
(function(){
const RAW=[[0.3,8.1],[0.7,9.8],[1.1,7.4],[1.6,11.2],[2.0,9.6],[2.5,13.1],
[3.0,10.8],[3.4,14.5],[3.9,11.9],[4.3,15.2],[4.7,12.6],[5.1,16.3],
[5.5,13.4],[5.9,17.1],[6.3,14.2],[6.7,18.0],[7.0,8.2],[7.4,9.5]];
const cv=document.getElementById('td-cv');
const W=680,H=300,DPR=window.devicePixelRatio||1;
cv.width=W*DPR;cv.height=H*DPR;cv.style.width=W+'px';cv.style.height=H+'px';
const ctx=cv.getContext('2d');ctx.scale(DPR,DPR);
const PAD={l:52,r:20,t:24,b:34};
const CW=W-PAD.l-PAD.r,CH=H-PAD.t-PAD.b;
const xMn=0,xMx=8,yMn=5,yMx=20;
const sx=x=>PAD.l+(x-xMn)/(xMx-xMn)*CW;
const sy=y=>PAD.t+CH-(y-yMn)/(yMx-yMn)*CH;
function rss(pts){
if(!pts.length)return 0;
const m=pts.reduce((s,p)=>s+p[1],0)/pts.length;
return pts.reduce((s,p)=>s+(p[1]-m)**2,0);
}
function buildTree(pts,depth){
if(depth===0||pts.length<2)return{leaf:true,val:pts.reduce((s,p)=>s+p[1],0)/(pts.length||1)};
let best=null,bestRSS=Infinity;
const xs=pts.map(p=>p[0]).sort((a,b)=>a-b);
for(let i=0;i<xs.length-1;i++){
const t=(xs[i]+xs[i+1])/2;
const L=pts.filter(p=>p[0]<=t),R=pts.filter(p=>p[0]>t);
if(!L.length||!R.length)continue;
const r=rss(L)+rss(R);
if(r<bestRSS){bestRSS=r;best={t,L,R};}
}
if(!best)return{leaf:true,val:pts.reduce((s,p)=>s+p[1],0)/pts.length};
return{leaf:false,split:best.t,left:buildTree(best.L,depth-1),right:buildTree(best.R,depth-1)};
}
function predict(tree,x){
if(tree.leaf)return tree.val;
return x<=tree.split?predict(tree.left,x):predict(tree.right,x);
}
function countLeaves(tree){
if(tree.leaf)return 1;
return countLeaves(tree.left)+countLeaves(tree.right);
}
function depthColor(d){
const t=Math.min((d-1)/6,1);
const r=Math.round(37+t*(220-37)),g=Math.round(150+t*(38-150)),b=Math.round(60+t*(38-60));
return`rgb(${r},${g},${b})`;
}
function draw(depth){
const tree=buildTree(RAW,depth);
const leaves=countLeaves(tree);
const trainRSS=RAW.reduce((s,p)=>s+(p[1]-predict(tree,p[0]))**2,0);
ctx.clearRect(0,0,W,H);
ctx.fillStyle='#f8f9fb';ctx.fillRect(PAD.l,PAD.t,CW,CH);
ctx.strokeStyle='#e8ecf3';ctx.lineWidth=0.7;
[2,4,6,8].forEach(x=>{ctx.beginPath();ctx.moveTo(sx(x),PAD.t);ctx.lineTo(sx(x),PAD.t+CH);ctx.stroke();});
[5,10,15,20].forEach(y=>{ctx.beginPath();ctx.moveTo(PAD.l,sy(y));ctx.lineTo(PAD.l+CW,sy(y));ctx.stroke();});
ctx.strokeStyle='#9aa3b5';ctx.lineWidth=1.2;
ctx.beginPath();ctx.moveTo(PAD.l,PAD.t);ctx.lineTo(PAD.l,PAD.t+CH);ctx.lineTo(PAD.l+CW,PAD.t+CH);ctx.stroke();
ctx.fillStyle='#888';ctx.font='10px Georgia,serif';ctx.textAlign='center';
[0,2,4,6,8].forEach(x=>ctx.fillText(x.toFixed(0),sx(x),PAD.t+CH+13));
ctx.textAlign='right';
[5,10,15,20].forEach(y=>ctx.fillText(y.toFixed(0),PAD.l-5,sy(y)+3));
const col=depthColor(depth);
const step=0.03;
ctx.strokeStyle=col;ctx.lineWidth=2.5;ctx.beginPath();
let first=true;
for(let x=xMn;x<=xMx;x+=step){
const yp=predict(tree,x);
if(first){ctx.moveTo(sx(x),sy(yp));first=false;}else ctx.lineTo(sx(x),sy(yp));
}
ctx.stroke();
RAW.forEach(p=>{
ctx.beginPath();ctx.arc(sx(p[0]),sy(p[1]),5,0,2*Math.PI);
ctx.fillStyle='#1a3a5c';ctx.fill();
ctx.strokeStyle='#fff';ctx.lineWidth=1.5;ctx.stroke();
});
ctx.fillStyle='#1a2e45';ctx.font='bold 11px Georgia,serif';ctx.textAlign='center';
ctx.fillText('Ajuste del árbol de regresión',PAD.l+CW/2,PAD.t-7);
const overfit=depth>=5?'⚠️ Posible sobreajuste':depth>=3?'Ajuste razonable':'Modelo simple';
document.getElementById('td-info').innerHTML=
`Profundidad = <strong>${depth}</strong> · `+
`Hojas = <strong>${leaves}</strong> · `+
`RSS entrenamiento = <strong>${trainRSS.toFixed(2)}</strong> · `+
`<span style="color:${depth>=5?'#dc2626':depth>=3?'#ca8a04':'#16a34a'}">${overfit}</span>`;
}
document.getElementById('td-sl').addEventListener('input',function(){
const d=parseInt(this.value);
document.getElementById('td-dv').textContent=d;
draw(d);
});
draw(2);
})();
</script>
```
## Evaluación de árboles de regresión
En regresión, una métrica frecuente es el coeficiente de determinación $R^2$. Este compara el error del modelo con el error de un modelo base que siempre predice el promedio de la variable respuesta.
::: {.callout-note appearance=“minimal”}
Definición. El coeficiente de determinación $R^2$ se define como:
$$
R^2=1-\frac{SSE}{SS_{TOT}},
$$
con
$$
SSE=\sum_i(y_i-\hat y_i)^2,
\qquad
SS_{TOT}=\sum_i(y_i-\bar y)^2.
$$ {#eq-r2}
:::
Un valor $R^2=0.90$ indica que el modelo explica aproximadamente el 90% de la variabilidad de la respuesta respecto al promedio. Un valor cercano a cero indica que el modelo no mejora sustancialmente frente a predecir siempre la media. En conjuntos de prueba, un $R^2$ negativo también es posible: significa que el modelo generaliza peor que el predictor promedio.
Además de $R^2$, se suelen reportar el error absoluto medio (MAE), el error cuadrático medio (MSE) y la raíz del error cuadrático medio (RMSE). La selección de métrica depende del problema. Si se desea penalizar mucho los errores grandes, RMSE puede ser adecuado; si se busca una medida más robusta ante valores extremos, MAE suele ser más interpretable.
## Ventajas y limitaciones de los árboles
Los árboles de regresión tienen varias ventajas. Son fáciles de entender porque se parecen a diagramas de decisión; producen reglas explícitas; pueden capturar relaciones no lineales e interacciones; requieren poca preparación de variables; y pueden trabajar con distintos criterios de error [@james2021introduction]. Algunos algoritmos de árboles también incorporan estrategias para tratar datos faltantes, como divisiones sustitutas o *surrogate splits* [@breiman1984classification].
Sus limitaciones también son importantes. Primero, tienden a sobreajustarse si crecen demasiado. Segundo, sus particiones rectangulares pueden ser ineficientes cuando la verdadera relación entre variables es suave u oblicua. Tercero, son inestables: pequeños cambios en los datos pueden producir árboles muy distintos. Cuarto, pueden ser sensibles al ruido y a valores atípicos. Finalmente, si llegan nuevos datos, normalmente se reentrena el árbol completo.
Estas debilidades motivan el uso de métodos de ensamble, donde se combinan muchos árboles para obtener predicciones más estables y precisas.
## Bagging: agregación por bootstrap
Bagging, abreviatura de *bootstrap aggregating*, fue propuesto por Breiman como una técnica para mejorar la estabilidad y precisión de predictores inestables [@breiman1996bagging]. La idea consiste en entrenar muchos modelos sobre muestras bootstrap del conjunto original y combinar sus predicciones.
::: {.callout-note appearance=“minimal”}
Definición. Una muestra bootstrap de tamaño $n$ se obtiene seleccionando $n$ observaciones con reemplazo a partir del conjunto de entrenamiento original.
$$
\mathcal{D}^{*(b)}=\{(x_1^{*(b)},y_1^{*(b)}),\ldots,(x_n^{*(b)},y_n^{*(b)})\}.
$$ {#eq-bootstrap-sample}
:::
El procedimiento general es:
1. Generar muchas muestras bootstrap del conjunto de entrenamiento.
2. Entrenar un árbol independiente en cada muestra.
3. Combinar las predicciones de todos los árboles.
En regresión, la combinación suele ser el promedio:
::: {.callout-note appearance=“minimal”}
Definición. La predicción bagging para regresión es el promedio de las predicciones de $B$ modelos entrenados sobre muestras bootstrap:
$$
\hat f_{bag}(x)=\frac{1}{B}\sum_{b=1}^{B}\hat f^{*(b)}(x).
$$ {#eq-bagging}
:::
Bagging reduce la varianza sin aumentar demasiado el sesgo. Es especialmente útil con árboles de decisión, porque los árboles individuales pueden variar mucho ante cambios pequeños en los datos.
```{python}
from sklearn.ensemble import BaggingRegressor
bag = BaggingRegressor(
estimator=DecisionTreeRegressor(random_state=42),
n_estimators=100,
random_state=42,
n_jobs=-1
)
bag.fit(X_train, y_train)
y_pred_bag = bag.predict(X_test)
print("MAE bagging:", mean_absolute_error(y_test, y_pred_bag))
print("R2 bagging:", r2_score(y_test, y_pred_bag))
```
## Random forests
Random forest extiende bagging introduciendo aleatoriedad adicional en la construcción de cada árbol. Además de entrenar cada árbol con una muestra bootstrap, en cada división se considera solo un subconjunto aleatorio de variables candidatas [@breiman2001random]. Esto disminuye la correlación entre árboles y mejora la reducción de varianza.
::: {.callout-note appearance=“minimal”}
Definición. Un random forest es un ensamble de árboles entrenados sobre muestras bootstrap, donde cada división se elige considerando solo un subconjunto aleatorio de variables.
$$
\hat f_{RF}(x)=\frac{1}{B}\sum_{b=1}^{B}\hat f_b(x).
$$ {#eq-random-forest}
:::
El modelo conserva parte de la interpretabilidad de los árboles, por ejemplo mediante medidas de importancia de variables, pero su estructura global ya no se resume en un único diagrama. A cambio, suele ofrecer mejor desempeño predictivo y menor inestabilidad.
```{python}
from sklearn.ensemble import RandomForestRegressor
rf = RandomForestRegressor(
n_estimators=300,
max_features="sqrt",
random_state=42,
n_jobs=-1
)
rf.fit(X_train, y_train)
y_pred_rf = rf.predict(X_test)
print("MAE random forest:", mean_absolute_error(y_test, y_pred_rf))
print("R2 random forest:", r2_score(y_test, y_pred_rf))
importances = pd.Series(rf.feature_importances_, index=X_train.columns)
importances.sort_values(ascending=False).head(10)
```
## Boosting
Boosting también combina varios modelos, pero a diferencia de bagging, los entrena de forma secuencial. Cada nuevo modelo intenta corregir los errores cometidos por los modelos anteriores. La intuición es construir un predictor fuerte a partir de muchos aprendices débiles [@freund1997decision; @schapire1990strength].
En clasificación, algunos métodos de boosting asignan mayor peso a las observaciones mal clasificadas. En regresión, una forma muy influyente es gradient boosting, donde cada nuevo árbol se ajusta a los residuos o, más generalmente, a la dirección de descenso del gradiente de una función de pérdida [@friedman2001greedy].
::: {.callout-note appearance=“minimal”}
Definición. En gradient boosting para regresión con pérdida cuadrática, el modelo se construye aditivamente:
$$
F_M(x)=F_0(x)+\sum_{m=1}^{M}\nu h_m(x),
$$
donde cada $h_m$ se ajusta a los residuos del modelo anterior y $\nu$ es la tasa de aprendizaje.
$$ {#eq-gradient-boosting}
:::
El procedimiento conceptual es:
1. Entrenar un primer modelo simple.
2. Calcular los errores o residuos.
3. Entrenar un nuevo modelo para explicar esos errores.
4. Repetir el proceso varias veces.
5. Combinar los modelos, usualmente con pesos o una tasa de aprendizaje.
Boosting puede alcanzar alta precisión, pero suele ser más sensible al ruido, a valores atípicos y al sobreajuste. Por ello requiere controlar hiperparámetros como número de estimadores, profundidad de los árboles, tasa de aprendizaje y regularización.
```{python}
from sklearn.ensemble import GradientBoostingRegressor
gb = GradientBoostingRegressor(
n_estimators=300,
learning_rate=0.05,
max_depth=3,
random_state=42
)
gb.fit(X_train, y_train)
y_pred_gb = gb.predict(X_test)
print("MAE gradient boosting:", mean_absolute_error(y_test, y_pred_gb))
print("R2 gradient boosting:", r2_score(y_test, y_pred_gb))
```
Entre las implementaciones modernas de boosting se encuentran XGBoost, LightGBM y CatBoost. Estas bibliotecas agregan optimizaciones computacionales y regularización avanzada, y son comunes en competencias y aplicaciones tabulares de alto rendimiento [@chen2016xgboost; @ke2017lightgbm; @prokhorenkova2018catboost].
## Bagging frente a boosting
Bagging y boosting combinan modelos, pero responden a problemas distintos.
| Característica | Bagging | Boosting |
|---|---|---|
| Entrenamiento | Modelos independientes | Modelos secuenciales |
| Objetivo principal | Reducir varianza | Reducir sesgo y mejorar precisión |
| Manejo de errores | Las muestras se tratan de forma similar | Se enfoca en errores previos |
| Paralelización | Naturalmente paralelizable | Menos paralelizable por dependencia secuencial |
| Ruido y atípicos | Más robusto | Más sensible |
| Ejemplos | Bagging, Random Forest | AdaBoost, Gradient Boosting, XGBoost, LightGBM, CatBoost |
Como regla práctica, bagging es recomendable cuando el modelo base tiene alta varianza y tendencia al sobreajuste, como ocurre con árboles profundos. Boosting es útil cuando se busca mayor precisión y el modelo base todavía subajusta patrones relevantes, aunque debe regularizarse cuidadosamente.
## Clasificación del vecino más cercano
El algoritmo del vecino más cercano pertenece a otra familia de métodos supervisados. En lugar de construir reglas explícitas como un árbol, almacena los datos de entrenamiento y clasifica un nuevo punto según las observaciones más parecidas. Por ello se considera un método basado en memoria o *lazy learning*: el “aprendizaje” ocurre principalmente al guardar los casos, mientras que el esfuerzo computacional se desplaza al momento de predecir [@cover1967nearest; @duda2001pattern].
Sea un conjunto de entrenamiento
$$
S_N=\{(x_1,\theta_1),(x_2,\theta_2),\ldots,(x_N,\theta_N)\},
$$
donde $x_i\in\mathbb{R}^p$ es el vector de atributos y $\theta_i\in\{1,\ldots,c\}$ es la etiqueta de clase. Dada una métrica o función de distancia $d(x,z)$, el clasificador de vecino más cercano busca el punto de entrenamiento más próximo a una nueva observación $x$.
::: {.callout-note appearance=“minimal”}
Definición. En el clasificador de vecino más cercano, una nueva observación $x$ recibe la clase de la observación de entrenamiento más cercana bajo una métrica de distancia $d$:
$$
\hat y(x)=\theta_{i^*},\qquad i^*=\arg\min_{i=1,\ldots,N} d(x,x_i).
$$ {#eq-nearest-neighbor}
:::
También puede formularse mediante funciones discriminantes. Para cada clase $\omega_j$, se calcula la distancia mínima entre $x$ y los ejemplos de esa clase; se asigna la clase cuya distancia mínima sea menor.
::: {.callout-note appearance=“minimal”}
Definición. El discriminante de distancia mínima para la clase $\omega_j$ es:
$$
g_j(x)=\min_{i:\theta_i=j} d(x,x_i),\qquad
\hat y(x)=\arg\min_{j=1,
\ldots,c} g_j(x).
$$ {#eq-nn-discriminant}
:::
Esta formulación muestra la conexión con los clasificadores por partes: si cada muestra de entrenamiento se trata como una subclase, entonces la regla de decisión se reduce a escoger la muestra más cercana.
## Métricas de distancia para vecinos cercanos
La elección de la distancia es crucial. KNN no “entiende” directamente el significado semántico de las variables; solo compara puntos mediante una métrica. Por eso, variables medidas en escalas grandes pueden dominar la decisión si no se normalizan o estandarizan. Además, distintas métricas inducen geometrías distintas: la distancia euclidiana produce vecindades esféricas, mientras que la distancia Manhattan produce vecindades con forma de rombo en dos dimensiones.
::: {.callout-note appearance=“minimal”}
Definición. La distancia de Minkowski de orden $s\geq 1$ entre $x,z\in\mathbb{R}^p$ se define como:
$$
d_s(x,z)=\left(\sum_{j=1}^{p}|x_j-z_j|^s\right)^{1/s}.
$$ {#eq-minkowski-distance}
:::
Casos particulares importantes son la distancia Manhattan ($s=1$) y la distancia euclidiana ($s=2$).
::: {.callout-note appearance=“minimal”}
Definición. La distancia Manhattan o taxicab entre $x$ y $z$ es:
$$
d_1(x,z)=\sum_{j=1}^{p}|x_j-z_j|.
$$ {#eq-manhattan-distance}
:::
::: {.callout-note appearance=“minimal”}
Definición. La distancia euclidiana entre $x$ y $z$ es:
$$
d_2(x,z)=\sqrt{\sum_{j=1}^{p}(x_j-z_j)^2}.
$$ {#eq-euclidean-distance}
:::
Otra opción es la distancia de Chebyshev, que mide la máxima diferencia coordenada a coordenada.
::: {.callout-note appearance=“minimal”}
Definición. La distancia de Chebyshev entre $x$ y $z$ es:
$$
d_{\infty}(x,z)=\max_{j=1,
\ldots,p}|x_j-z_j|.
$$ {#eq-chebyshev-distance}
:::
También se usan distancias cuadráticas ponderadas, útiles cuando las variables tienen distinta escala o correlación. Si $Q$ es una matriz simétrica definida positiva, entonces
::: {.callout-note appearance=“minimal”}
Definición. Una distancia cuadrática ponderada entre $x$ y $z$ puede escribirse como:
$$
d_Q(x,z)=(x-z)^{\top}Q(x-z).
$$ {#eq-quadratic-distance}
:::
Un caso relevante es la distancia de Mahalanobis, donde $Q=\Sigma^{-1}$ y $\Sigma$ es la matriz de covarianza. Esta distancia corrige por escala y correlación entre variables [@duda2001pattern].
```{python}
import numpy as np
x = np.array([30, 25])
z = np.array([28, 24])
manhattan = np.sum(np.abs(x - z))
euclidean = np.sqrt(np.sum((x - z)**2))
chebyshev = np.max(np.abs(x - z))
print({
"Manhattan": manhattan,
"Euclidiana": euclidean,
"Chebyshev": chebyshev
})
```
```{=html}
<div id="mk-wrap" style="font-family:Georgia,serif;max-width:700px;margin:2em auto;background:#f9fafc;border:1px solid #d0d7e3;border-radius:10px;padding:20px 24px;box-shadow:0 2px 12px rgba(30,60,120,.07);">
<div style="text-align:center;margin-bottom:12px;">
<span style="font-size:1.05em;font-weight:bold;color:#1a2e45;">Bola unitaria de Minkowski — explorador interactivo</span><br>
<span style="font-size:0.81em;color:#666;">Arrastra p para ver cómo cambia la forma de la bola: diamante (p=1), círculo (p=2), cuadrado (p→∞)</span>
</div>
<div style="display:flex;align-items:center;gap:12px;margin-bottom:10px;flex-wrap:wrap;">
<label style="font-size:0.83em;color:#444;">p = <strong id="mk-pv">2</strong>  <span id="mk-name" style="color:#1a3a5c;font-style:italic;"></span></label>
<input id="mk-sl" type="range" min="1" max="20" step="1" value="2"
style="flex:1;min-width:150px;accent-color:#1a3a5c;">
</div>
<canvas id="mk-cv" style="display:block;width:100%;border-radius:6px;border:1px solid #e0e5ed;background:#fff;"></canvas>
</div>
<script>
(function(){
const cv=document.getElementById('mk-cv');
const W=680,H=360,DPR=window.devicePixelRatio||1;
cv.width=W*DPR;cv.height=H*DPR;cv.style.width=W+'px';cv.style.height=H+'px';
const ctx=cv.getContext('2d');ctx.scale(DPR,DPR);
const CX=W/2,CY=H/2,SCALE=140;
function ballR(theta,p){
const ac=Math.abs(Math.cos(theta)),as=Math.abs(Math.sin(theta));
if(p>=18)return 1/Math.max(ac,as);
return 1/Math.pow(Math.pow(ac,p)+Math.pow(as,p),1/p);
}
function drawBall(p,col,alpha,lw){
ctx.beginPath();
const N=720;
for(let i=0;i<=N;i++){
const th=i/N*2*Math.PI;
const r=ballR(th,p);
const x=CX+r*Math.cos(th)*SCALE,y=CY-r*Math.sin(th)*SCALE;
i===0?ctx.moveTo(x,y):ctx.lineTo(x,y);
}
ctx.closePath();
ctx.strokeStyle=col;ctx.globalAlpha=alpha;ctx.lineWidth=lw;ctx.stroke();
ctx.globalAlpha=1;
}
function draw(p){
ctx.clearRect(0,0,W,H);
ctx.fillStyle='#f8f9fb';ctx.fillRect(0,0,W,H);
ctx.strokeStyle='#e8ecf3';ctx.lineWidth=0.8;
[-1,1].forEach(v=>{
ctx.beginPath();ctx.moveTo(CX+v*SCALE,CY-SCALE*1.35);ctx.lineTo(CX+v*SCALE,CY+SCALE*1.35);ctx.stroke();
ctx.beginPath();ctx.moveTo(CX-SCALE*1.35,CY+v*SCALE);ctx.lineTo(CX+SCALE*1.35,CY+v*SCALE);ctx.stroke();
});
ctx.strokeStyle='#c9d0de';ctx.lineWidth=1;
ctx.beginPath();ctx.moveTo(CX-SCALE*1.35,CY);ctx.lineTo(CX+SCALE*1.35,CY);ctx.stroke();
ctx.beginPath();ctx.moveTo(CX,CY-SCALE*1.35);ctx.lineTo(CX,CY+SCALE*1.35);ctx.stroke();
ctx.fillStyle='#aaa';ctx.font='10px Georgia,serif';ctx.textAlign='center';
ctx.fillText('1',CX+SCALE,CY+13);ctx.fillText('-1',CX-SCALE,CY+13);
ctx.fillText('1',CX+6,CY-SCALE+5);ctx.fillText('-1',CX+8,CY+SCALE+5);
drawBall(1,'#dc2626',0.35,1.5);
drawBall(2,'#16a34a',0.35,1.5);
ctx.fillStyle='#dc2626';ctx.font='10px Georgia,serif';ctx.textAlign='left';
ctx.globalAlpha=0.55;ctx.fillText('p=1 (Manhattan)',CX+SCALE*0.72+4,CY-SCALE*0.12);ctx.globalAlpha=1;
ctx.fillStyle='#16a34a';
ctx.globalAlpha=0.55;ctx.fillText('p=2 (Euclídea)',CX+SCALE*0.72+4,CY-SCALE*0.12+14);ctx.globalAlpha=1;
const col=p<=1?'#dc2626':p<=2?'#2563eb':p<=5?'#7c3aed':p>=18?'#0f172a':'#1a3a5c';
ctx.fillStyle=col+'22';
ctx.beginPath();
const N=720;
for(let i=0;i<=N;i++){
const th=i/N*2*Math.PI;
const r=ballR(th,p);
const x=CX+r*Math.cos(th)*SCALE,y=CY-r*Math.sin(th)*SCALE;
i===0?ctx.moveTo(x,y):ctx.lineTo(x,y);
}
ctx.closePath();ctx.fill();
drawBall(p,col,0.9,2.5);
ctx.fillStyle='#1a2e45';ctx.font='bold 11px Georgia,serif';ctx.textAlign='center';
ctx.fillText('Bola unitaria Minkowski (p='+p+')',CX,22);
const name=p<=1?'Distancia Manhattan (L¹)':p===2?'Distancia Euclídea (L²)':p>=18?'Distancia Chebyshev (L∞)':'Distancia Minkowski (L'+p+')';
document.getElementById('mk-name').textContent=name;
}
document.getElementById('mk-sl').addEventListener('input',function(){
const p=parseInt(this.value);
document.getElementById('mk-pv').textContent=p>=18?'∞':p;
draw(p);
});
draw(2);
document.getElementById('mk-name').textContent='Distancia Euclídea (L²)';
})();
</script>
```
## De 1-NN a k-NN
La regla 1-NN puede generar regiones de clasificación muy fragmentadas, pues una sola observación ruidosa puede modificar la frontera local. Para reducir esa sensibilidad, se generaliza el método considerando los $k$ vecinos más cercanos. En clasificación, cada vecino vota por su clase; en regresión, se promedian sus respuestas.
::: {.callout-note appearance=“minimal”}
Definición. En $k$-NN para clasificación, si $N_k(x)$ es el conjunto de índices de los $k$ vecinos más cercanos de $x$, el discriminante de la clase $j$ es el número de vecinos que pertenecen a esa clase:
$$
g_j(x)=\sum_{i\in N_k(x)}\mathbb{I}(\theta_i=j),\qquad
\hat y(x)=\arg\max_{j=1,
\ldots,c}g_j(x).
$$ {#eq-knn-classification}
:::
::: {.callout-note appearance=“minimal”}
Definición. En $k$-NN para regresión, la predicción es el promedio de las respuestas de los $k$ vecinos más cercanos:
$$
\hat f(x)=\frac{1}{k}\sum_{i\in N_k(x)}y_i.
$$ {#eq-knn-regression}
:::
La elección de $k$ controla el equilibrio sesgo-varianza. Con $k=1$, el modelo tiene bajo sesgo local, pero alta varianza: puede adaptarse demasiado a puntos individuales. Con un $k$ grande, la frontera se suaviza y la varianza disminuye, pero aumenta el sesgo porque se promedian observaciones que pueden pertenecer a regiones distintas [@hastie2009elements]. En aplicaciones prácticas, $k$ suele elegirse mediante validación cruzada. Para clasificación binaria, a menudo se usan valores impares de $k$ para reducir empates.
```{python}
from sklearn.model_selection import train_test_split, GridSearchCV
from sklearn.neighbors import KNeighborsClassifier
from sklearn.preprocessing import StandardScaler
from sklearn.pipeline import Pipeline
from sklearn.metrics import accuracy_score, classification_report
import pandas as pd
clientes = pd.DataFrame({
"edad": [25, 28, 32, 35, 29, 31, 40, 22],
"ingreso": [20, 24, 26, 30, 23, 27, 35, 18],
"compra": ["No compra", "No compra", "Compra", "Compra", "No compra", "Compra", "Compra", "No compra"]
})
X_cls = clientes[["edad", "ingreso"]]
y_cls = clientes["compra"]
knn = Pipeline([
("scaler", StandardScaler()),
("model", KNeighborsClassifier())
])
param_grid = {"model__n_neighbors": [1, 3, 5]}
search = GridSearchCV(knn, param_grid=param_grid, cv=2)
search.fit(X_cls, y_cls)
nuevo_cliente = pd.DataFrame({"edad": [30], "ingreso": [25]})
print("Mejor k:", search.best_params_)
print("Predicción:", search.predict(nuevo_cliente))
```
```{=html}
<div id="knn-wrap" style="font-family:Georgia,serif;max-width:700px;margin:2em auto;background:#f9fafc;border:1px solid #d0d7e3;border-radius:10px;padding:20px 24px;box-shadow:0 2px 12px rgba(30,60,120,.07);">
<div style="text-align:center;margin-bottom:12px;">
<span style="font-size:1.05em;font-weight:bold;color:#1a2e45;">k-NN clasificador — explorador interactivo</span><br>
<span style="font-size:0.81em;color:#666;">Haz clic en el canvas para consultar un punto; ajusta k para ver cómo cambia la frontera de decisión</span>
</div>
<div style="display:flex;align-items:center;gap:12px;margin-bottom:10px;flex-wrap:wrap;">
<label style="font-size:0.83em;color:#444;">k = <strong id="knn-kv">3</strong></label>
<input id="knn-sl" type="range" min="1" max="15" step="1" value="3"
style="flex:1;min-width:150px;accent-color:#1a3a5c;">
</div>
<div id="knn-info" style="text-align:center;font-size:0.82em;padding:5px 10px;background:#fff;border:1px solid #e0e5ed;border-radius:5px;margin-bottom:10px;min-height:22px;">Haz clic en el canvas para clasificar un punto</div>
<canvas id="knn-cv" style="display:block;width:100%;border-radius:6px;border:1px solid #e0e5ed;background:#fff;cursor:crosshair;"></canvas>
</div>
<script>
(function(){
const TRAIN=[
{x:[1.2,3.8],y:'A'},{x:[1.8,4.5],y:'A'},{x:[2.3,3.2],y:'A'},{x:[1.5,5.1],y:'A'},
{x:[2.8,4.8],y:'A'},{x:[3.1,3.6],y:'A'},{x:[0.9,4.2],y:'A'},{x:[2.0,5.5],y:'A'},
{x:[3.5,5.0],y:'A'},{x:[1.3,2.9],y:'A'},{x:[2.6,4.1],y:'A'},{x:[3.8,4.4],y:'A'},
{x:[4.5,1.2],y:'B'},{x:[5.0,0.8],y:'B'},{x:[5.8,2.1],y:'B'},{x:[4.2,1.8],y:'B'},
{x:[6.1,1.5],y:'B'},{x:[5.5,2.8],y:'B'},{x:[4.8,0.5],y:'B'},{x:[6.5,2.5],y:'B'},
{x:[5.2,1.0],y:'B'},{x:[4.0,2.5],y:'B'},{x:[6.8,1.0],y:'B'},{x:[5.7,3.2],y:'B'}
];
const cv=document.getElementById('knn-cv');
const W=680,H=380,DPR=window.devicePixelRatio||1;
cv.width=W*DPR;cv.height=H*DPR;cv.style.width=W+'px';cv.style.height=H+'px';
const ctx=cv.getContext('2d');ctx.scale(DPR,DPR);
const PAD={l:44,r:16,t:16,b:30};
const CW=W-PAD.l-PAD.r,CH=H-PAD.t-PAD.b;
const xMn=0,xMx=8,yMn=0,yMx=7;
const sx=x=>PAD.l+(x-xMn)/(xMx-xMn)*CW;
const sy=y=>PAD.t+CH-(y-yMn)/(yMx-yMn)*CH;
const ix=px=>(px-PAD.l)/CW*(xMx-xMn)+xMn;
const iy=py=>yMx-(py-PAD.t)/CH*(yMx-yMn);
const COL={A:'#2563eb',B:'#dc2626'};
let query=null,k=3;
function dist(a,bq){return Math.sqrt((a.x[0]-bq[0])**2+(a.x[1]-bq[1])**2);}
function knn(q,k){
const ranked=[...TRAIN].map(t=>({...t,d:dist(t,[q.x,q.y])})).sort((a,b)=>a.d-b.d);
const nbrs=ranked.slice(0,k);
const votes={A:0,B:0};nbrs.forEach(n=>votes[n.y]++);
const pred=votes.A>=votes.B?'A':'B';
return{nbrs,pred,votes};
}
function drawBg(k){
const step=12;
for(let px=PAD.l;px<PAD.l+CW;px+=step){
for(let py=PAD.t;py<PAD.t+CH;py+=step){
const qx=ix(px),qy=iy(py);
const {pred}=knn({x:qx,y:qy},k);
ctx.fillStyle=pred==='A'?'rgba(37,99,235,0.08)':'rgba(220,38,38,0.08)';
ctx.fillRect(px,py,step,step);
}
}
}
function draw(){
ctx.clearRect(0,0,W,H);
drawBg(k);
ctx.strokeStyle='#e8ecf3';ctx.lineWidth=0.7;
[0,2,4,6,8].forEach(x=>{ctx.beginPath();ctx.moveTo(sx(x),PAD.t);ctx.lineTo(sx(x),PAD.t+CH);ctx.stroke();});
[0,2,4,6].forEach(y=>{ctx.beginPath();ctx.moveTo(PAD.l,sy(y));ctx.lineTo(PAD.l+CW,sy(y));ctx.stroke();});
ctx.strokeStyle='#9aa3b5';ctx.lineWidth=1.2;
ctx.beginPath();ctx.moveTo(PAD.l,PAD.t);ctx.lineTo(PAD.l,PAD.t+CH);ctx.lineTo(PAD.l+CW,PAD.t+CH);ctx.stroke();
ctx.fillStyle='#888';ctx.font='10px Georgia,serif';ctx.textAlign='center';
[0,2,4,6,8].forEach(x=>ctx.fillText(x,sx(x),PAD.t+CH+13));
ctx.textAlign='right';
[0,2,4,6].forEach(y=>ctx.fillText(y,PAD.l-5,sy(y)+3));
let result=null;
if(query){
result=knn(query,k);
const kDist=result.nbrs[result.nbrs.length-1].d;
ctx.beginPath();ctx.arc(sx(query.x),sy(query.y),kDist/(xMx-xMn)*CW,0,2*Math.PI);
ctx.strokeStyle='rgba(100,100,100,0.35)';ctx.lineWidth=1.5;ctx.setLineDash([5,4]);ctx.stroke();ctx.setLineDash([]);
result.nbrs.forEach((n,i)=>{
ctx.strokeStyle=COL[n.y]+'99';ctx.lineWidth=1.2;
ctx.beginPath();ctx.moveTo(sx(query.x),sy(query.y));ctx.lineTo(sx(n.x[0]),sy(n.x[1]));ctx.stroke();
ctx.fillStyle=COL[n.y];ctx.font='bold 9px Georgia,serif';ctx.textAlign='center';
const mx=(sx(query.x)+sx(n.x[0]))/2,my=(sy(query.y)+sy(n.x[1]))/2;
ctx.fillText(i+1,mx,my);
});
}
TRAIN.forEach(t=>{
ctx.beginPath();ctx.arc(sx(t.x[0]),sy(t.x[1]),6,0,2*Math.PI);
ctx.fillStyle=COL[t.y]+'cc';ctx.fill();
ctx.strokeStyle='#fff';ctx.lineWidth=1.5;ctx.stroke();
ctx.fillStyle=COL[t.y];ctx.font='bold 9px Georgia,serif';ctx.textAlign='center';
ctx.fillText(t.y,sx(t.x[0]),sy(t.x[1])+3);
});
if(query){
const col=result?COL[result.pred]:'#666';
const S=sx(query.x),Sy=sy(query.y),R=9;
ctx.beginPath();
ctx.moveTo(S,Sy-R);ctx.lineTo(S+R*0.35,Sy-R*0.35);ctx.lineTo(S+R,Sy);
ctx.lineTo(S+R*0.35,Sy+R*0.35);ctx.lineTo(S,Sy+R);
ctx.lineTo(S-R*0.35,Sy+R*0.35);ctx.lineTo(S-R,Sy);
ctx.lineTo(S-R*0.35,Sy-R*0.35);ctx.closePath();
ctx.fillStyle=col;ctx.fill();ctx.strokeStyle='#fff';ctx.lineWidth=2;ctx.stroke();
if(result){
document.getElementById('knn-info').innerHTML=
`Consulta (${query.x.toFixed(2)}, ${query.y.toFixed(2)}) → `+
`<strong style="color:${col}">Clase ${result.pred}</strong> · `+
`Votos: A=${result.votes.A}, B=${result.votes.B} · `+
`k=${k} vecinos más cercanos`;
}
}
}
cv.addEventListener('click',function(e){
const rect=cv.getBoundingClientRect();
const px=e.clientX-rect.left,py=e.clientY-rect.top;
query={x:ix(px),y:iy(py)};
draw();
});
document.getElementById('knn-sl').addEventListener('input',function(){
k=parseInt(this.value);
document.getElementById('knn-kv').textContent=k;
draw();
});
draw();
})();
</script>
```
## Error asintótico de vecinos cercanos
Una razón histórica de la importancia de NN es que posee garantías teóricas simples. En el límite de muchas observaciones independientes e idénticamente distribuidas, el error asintótico de 1-NN está acotado por el error de Bayes, que es el menor error posible si se conocieran las distribuciones verdaderas de las clases [@cover1967nearest].
::: {.callout-note appearance=“minimal”}
Definición. Si $P^*$ es el error de Bayes y $P_1$ es el error asintótico de 1-NN con $c$ clases, entonces:
$$
P^*\leq P_1\leq P^*\left(2-\frac{c}{c-1}P^*\right).
$$ {#eq-cover-hart-bound}
:::
Como consecuencia, para dos o más clases se suele resumir la cota como $P_1\leq 2P^*$. Es decir, bajo los supuestos del resultado, 1-NN no es arbitrariamente malo en el límite: su error asintótico es, a lo más, aproximadamente el doble del error óptimo de Bayes.
Para $k$-NN, si $N\to\infty$, $k\to\infty$ y $k/N\to 0$, el método puede aproximar el rendimiento óptimo de Bayes. La condición $k/N\to 0$ expresa que el vecindario debe crecer en número de puntos, pero seguir siendo local en relación con el tamaño total de la muestra [@stone1977consistent; @hastie2009elements].
::: {.callout-note appearance=“minimal”}
Definición. Una condición clásica de consistencia para $k$-NN es:
$$
N\to\infty,\qquad k\to\infty,\qquad \frac{k}{N}\to 0.
$$ {#eq-knn-consistency-condition}
:::
Estas conclusiones dependen de que los vecinos cercanos sean realmente cercanos en el espacio de atributos. En alta dimensión, esta condición puede fallar: incluso el vecino más cercano puede quedar lejos en términos geométricos. Este fenómeno forma parte de la llamada maldición de la dimensionalidad.
## Maldición de la dimensionalidad
KNN funciona mejor cuando la dimensión efectiva del problema no es demasiado alta y existe una noción de cercanía significativa. En espacios de alta dimensión, las distancias tienden a concentrarse: la diferencia entre el vecino más cercano y el más lejano puede volverse pequeña en relación con la escala total. Como resultado, la noción de “vecino” pierde poder discriminativo y se requieren muchas más muestras para cubrir el espacio [@hastie2009elements; @bellman1961adaptive].
::: {.callout-note appearance=“minimal”}
Definición. En un hipercubo unitario de dimensión $p$, una región con fracción lineal $r$ en cada coordenada ocupa una fracción de volumen:
$$
V(r,p)=r^p.
$$ {#eq-curse-volume}
:::
Esta expresión muestra por qué la dimensión importa. Si $r=0.1$ y $p=10$, el volumen relativo es $10^{-10}$. Para cubrir localmente un espacio de alta dimensión se necesitaría una cantidad enorme de datos. Por ello, antes de aplicar KNN conviene considerar selección de variables, reducción de dimensionalidad, escalamiento adecuado y validación cuidadosa.
## Métodos mejorados para vecinos cercanos
El método NN básico tiene tres limitaciones principales: debe almacenar todos los datos, debe comparar una consulta contra muchas observaciones y es sensible al ruido. Existen variantes que atacan estas dificultades.
### Búsqueda eficiente: branch and bound
Los algoritmos de búsqueda rápida reducen el tiempo de predicción mediante preparación fuera de línea. La idea general es organizar las muestras en una estructura jerárquica y descartar regiones completas cuando no pueden contener al vecino más cercano. Esta lógica aparece en métodos *branch and bound*, árboles k-d y árboles de bolas [@friedman1977algorithm; @fukunaga1975branch].
Supongamos que un nodo $p$ contiene un subconjunto de muestras $\mathcal{X}_p$, con centro $M_p$ y radio $r_p$ definido como la máxima distancia desde el centro a una muestra del nodo.
::: {.callout-note appearance=“minimal”}
Definición. Para un nodo $p$ con muestras $\mathcal{X}_p$, su centro y radio pueden definirse como:
$$
M_p=\frac{1}{N_p}\sum_{x_i\in\mathcal{X}_p}x_i,
\qquad
r_p=\max_{x_i\in\mathcal{X}_p}d(x_i,M_p).
$$ {#eq-node-center-radius}
:::
Si $B$ es la mejor distancia encontrada hasta el momento para una consulta $x$, se puede descartar un nodo cuando, por desigualdad triangular, ningún punto del nodo puede mejorar esa distancia. Una forma intuitiva de la regla es: si el centro del nodo está demasiado lejos de $x$ en comparación con su radio, no vale la pena explorar ese nodo.
::: {.callout-note appearance=“minimal”}
Definición. Una regla de descarte por cota inferior para un nodo $p$ es:
$$
d(x,M_p)-r_p>B.
$$ {#eq-branch-bound-prune}
:::
Si se cumple @eq-branch-bound-prune, entonces incluso el punto más favorable dentro del nodo estaría a una distancia mayor que $B$, por lo que el nodo puede descartarse.
### Edición de vecinos cercanos
La edición de vecinos cercanos busca eliminar muestras ambiguas o ruidosas, especialmente en regiones donde las clases se traslapan. La intuición es que ciertos puntos mal etiquetados o ubicados en zonas de conflicto pueden fragmentar la frontera de decisión y perjudicar la generalización [@wilson1972asymptotic].
Un procedimiento típico consiste en preclasificar cada muestra usando sus vecinos; si una muestra es clasificada incorrectamente por el resto del conjunto, se marca como candidata a eliminar. Este proceso puede repetirse varias rondas si se cuenta con suficientes datos. La edición puede mejorar robustez, aunque también puede eliminar casos difíciles pero legítimos, por lo que debe evaluarse con validación.
::: {.callout-note appearance=“minimal”}
Definición. La edición por preclasificación elimina una muestra $(x_i,y_i)$ si su etiqueta no coincide con la predicción obtenida al clasificarla usando el conjunto de entrenamiento sin esa muestra:
$$
(x_i,y_i)\text{ se elimina si } \hat y_{-i}(x_i)\neq y_i.
$$ {#eq-edited-nn-rule}
:::
### Condensed nearest neighbor
El método *condensed nearest neighbor* busca reducir memoria y cómputo conservando solo un subconjunto representativo de muestras [@hart1968condensed]. Se separa el conjunto original en dos partes: un conjunto almacenado $\mathcal{S}$ y un conjunto restante $\mathcal{G}$. Se comienza con pocas muestras en $\mathcal{S}$; luego se revisan las muestras de $\mathcal{G}$. Si una muestra se clasifica correctamente usando $\mathcal{S}$, no se agrega; si se clasifica mal, se incorpora a $\mathcal{S}$. Al final, solo $\mathcal{S}$ se usa para clasificar.
::: {.callout-note appearance=“minimal”}
Definición. El conjunto condensado $\mathcal{S}$ busca ser un subconjunto del entrenamiento tal que el clasificador 1-NN construido con $\mathcal{S}$ preserve, aproximadamente, las decisiones obtenidas con el conjunto completo:
$$
\mathcal{S}\subseteq S_N,
\qquad
\hat y_{\mathcal{S}}(x_i)\approx \hat y_{S_N}(x_i).
$$ {#eq-condensed-nn}
:::
```{python}
from sklearn.neighbors import KNeighborsClassifier
from sklearn.datasets import make_moons
from sklearn.model_selection import cross_val_score
from sklearn.pipeline import Pipeline
from sklearn.preprocessing import StandardScaler
X_demo, y_demo = make_moons(n_samples=300, noise=0.25, random_state=42)
scores = []
for k in [1, 3, 5, 11, 21, 51]:
model = Pipeline([
("scaler", StandardScaler()),
("knn", KNeighborsClassifier(n_neighbors=k))
])
acc = cross_val_score(model, X_demo, y_demo, cv=5, scoring="accuracy").mean()
scores.append({"k": k, "accuracy_cv": acc})
pd.DataFrame(scores)
```
## Cuándo usar KNN
KNN es una buena opción cuando el tamaño de muestra es razonable, la dimensión no es demasiado alta, las variables pueden escalarse de forma significativa y se desea una decisión basada en casos similares. Su interpretación es natural: “este punto se clasifica así porque se parece a estos ejemplos”. Sin embargo, no es ideal cuando hay millones de observaciones, demasiadas variables irrelevantes, datos muy ruidosos o necesidad de predicción en tiempo real sin estructuras de búsqueda especializadas.
## Comparación conceptual: árboles y vecinos cercanos
Los árboles y KNN son métodos no lineales, pero su lógica es distinta. Los árboles construyen particiones explícitas mediante reglas jerárquicas; KNN define vecindades alrededor del punto a predecir. Los árboles generan modelos relativamente rápidos al momento de predecir, porque basta recorrer una ruta desde la raíz hasta una hoja. KNN puede ser más costoso en predicción, porque debe calcular distancias con respecto a muchos puntos de entrenamiento, salvo que se usen estructuras de búsqueda eficientes.
Desde el punto de vista pedagógico, ambos métodos ayudan a entender que el aprendizaje supervisado no se reduce a ajustar rectas. Un modelo puede aproximar una función mediante regiones constantes, mediante promedios locales o mediante combinaciones de muchos modelos simples.
## Buenas prácticas de modelado
Para usar árboles, ensambles o KNN en un flujo reproducible de ciencia de datos, conviene seguir algunas reglas:
1. Separar datos de entrenamiento, validación y prueba, o usar validación cruzada.
2. Comparar contra modelos base simples, como predecir siempre el promedio en regresión.
3. Reportar métricas adecuadas al problema, no solo una métrica global.
4. Controlar hiperparámetros con validación, no con el conjunto de prueba.
5. Documentar semillas aleatorias, versiones de paquetes y criterios de evaluación.
6. Interpretar el modelo considerando sus supuestos geométricos: regiones rectangulares en árboles, distancias en KNN y reducción de varianza o sesgo en ensambles.
El siguiente ejemplo muestra una comparación breve entre árbol individual, random forest y gradient boosting.
```{python}
from sklearn.model_selection import cross_validate
models = {
"Árbol": DecisionTreeRegressor(random_state=42),
"Random Forest": RandomForestRegressor(n_estimators=200, random_state=42, n_jobs=-1),
"Gradient Boosting": GradientBoostingRegressor(random_state=42)
}
results = []
for name, model in models.items():
cv = cross_validate(
model,
X,
y,
cv=5,
scoring={"r2": "r2", "neg_mae": "neg_mean_absolute_error"},
n_jobs=-1
)
results.append({
"modelo": name,
"R2_promedio": cv["test_r2"].mean(),
"MAE_promedio": -cv["test_neg_mae"].mean()
})
pd.DataFrame(results).sort_values("R2_promedio", ascending=False)
```
## Conclusiones
Los árboles de regresión son modelos supervisados que reemplazan una ecuación global por una colección de reglas locales. Su predicción es constante dentro de cada región y se calcula como el promedio de las observaciones que caen en la hoja correspondiente. El entrenamiento se basa en particionamiento recursivo: se prueban cortes y se elige aquel que reduce más el error, usualmente medido como suma de cuadrados.
Su principal fortaleza es la interpretabilidad, pero también presentan riesgos: sobreajuste, sensibilidad al ruido, inestabilidad y fronteras rectangulares. La poda y el criterio de costo-complejidad ayudan a controlar árboles individuales. Los métodos de ensamble, como bagging, random forests y boosting, buscan superar estas limitaciones combinando muchos árboles. Bagging reduce varianza mediante entrenamiento paralelo sobre muestras bootstrap; boosting entrena modelos secuencialmente para corregir errores y suele obtener alta precisión bajo una buena regularización.
Finalmente, el vecino más cercano muestra otra forma de razonamiento no lineal: predecir a partir de casos similares. Mientras los árboles dividen el espacio mediante reglas, KNN usa distancias. En conjunto, estos métodos ofrecen una introducción poderosa al modelado predictivo moderno y preparan el terreno para técnicas más avanzadas de aprendizaje automático.